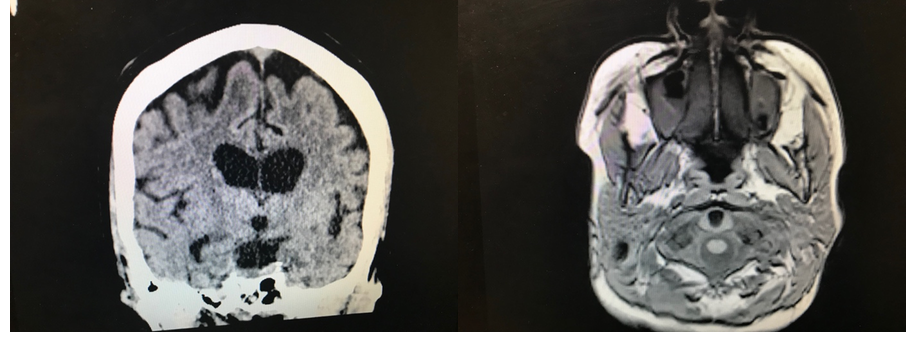
Case Presentation: A 52-year-old woman presented following a two-month history of bizarre behavior and altered mental status. Prior to the onset of symptoms, the patient was at a normal level of neurological functioning, though she did have a longstanding history of alcohol abuse with associated alcoholic hepatitis, liver steatosis, and protein-calorie malnutrition. Her past medical history also included stable pulmonary sarcoidosis and hypothyroidism.She was initially brought to medical attention when she was witnessed to have a seizure at a rehabilitation facility where she was staying following participation in an alcohol detoxification program. There she continued to have partial complex seizures with myoclonic extremity movements as well as one 40-minute episode of status epilepticus which lead to sedation and intubation in the ICU. After returning to the floor, she was observed to have waxing and waning episodes of confusion as well as apparent hallucinations, with associated agitation, paranoia, and aggressive overtures towards the staff and herself. She remained disoriented and aphasic, repeating stereotyped phrases. Myoclonus was noted of the right upper extremity. MRI showed signal involving the left temporal, parietal, and occipital lobes with extension towards the left hippocampus with surrounding edema.
Inpatient workup for her seizures during this two-week period revealed a head CT with no acute intracranial abnormalities. Laboratory work was significant only for mild macrocytic anemia, intermittent hypernatremia, and elevated CK of 304. TSH was within normal limits. Treponemal and HIV testing were negative. LP showed RBC 81, 56% PMNs, glucose 56, LDH 2.7, protein 28, no microorganisms, and myelin basic protein elevated at 6.5. CSF was unremarkable. She was maintained in the hospital on levetiracetam, thiamine, and folate with minimal improvement in her mental status for nine weeks. CJD protein revealed the presence of 14-3-3 protein, T-tau elevated at >4000 pg/mL, and negative RT-Qu1C. Her case including imaging studies was reviewed by the National Prion Disease Surveillance Center, who confirmed the diagnosis of Creutzfeldt-Jakob disease.
Discussion: Human prion disease occurs in most of the developed world at a rate of 1 to 1.5 per 1 million. In the United States, the incidence is around 1.2 in 1 million. Definitive diagnosis of CJD is performed through histopathological analysis, which can show spongiform brain degeneration, astrocytic gliosis and neuronal loss due to the accumulation of abnormal prion protein (PrPSc). Due to the rare incidence of CJD, its diagnosis is often not entertained at the time of presentation. Considering the inherent difficulties of performing pathological diagnosis and accessing the accuracy of lab markers, MRI became an important instrument in the evaluation of patients with suspected prion disease
Conclusions: Patients who have progressive dementia and associated atypical features should be investigated especially with DW MRI for CJD. According to diagnostic criteria of WHO for the probable diagnosis of CDJ, the presence of at least one criterion among typical EEG findings and 14-3-3 positivity for CSF samples or at least 2 criteria among myoclonus, visual disturbances, cerebellar, pyramidal or extrapyramidal findings and akinetic mutism together with progressive dementia are required. Cortical ribboning is a very useful diagnostic sign for CJD.