Introduction: Pheochromocytoma is a rare adrenal tumor causing hypertensive crisis. The classic triad of symptoms in patients include palpitations, headaches and tremor, however, not every patient manifests with this typical presentation. Here we present a case of pheochromoctyoma who had adrenergic crisis after receiving antihistamine medication in the emergency room.
Case Presentation: A 45 year old female with history of anxiety and migraines presented to the emergency room with nausea and headache. The initial workup in the emergency room was unremarkable, and the patient was given metoclopramide and diphenhydramine for symptom relief. Shortly after these medications were administered the patient became tachycardic, hypertensive, and hypoxemic. A stat chest x-ray revealed flash pulmonary edema and the patient was intubated for respiratory distress.
A complete metabolic panel and complete blood count were all within normal limits. EKG showed sinus bradycardia and shortened PR interval. Upon review of outside records, it was noted that the patient had a history of cardiac arrest during a prior surgery. She also had a history of transient reductions in systolic heart function with pulmonary edema that occurred perioperativley–subsequent echocardiograms showed a return of normal systolic function.
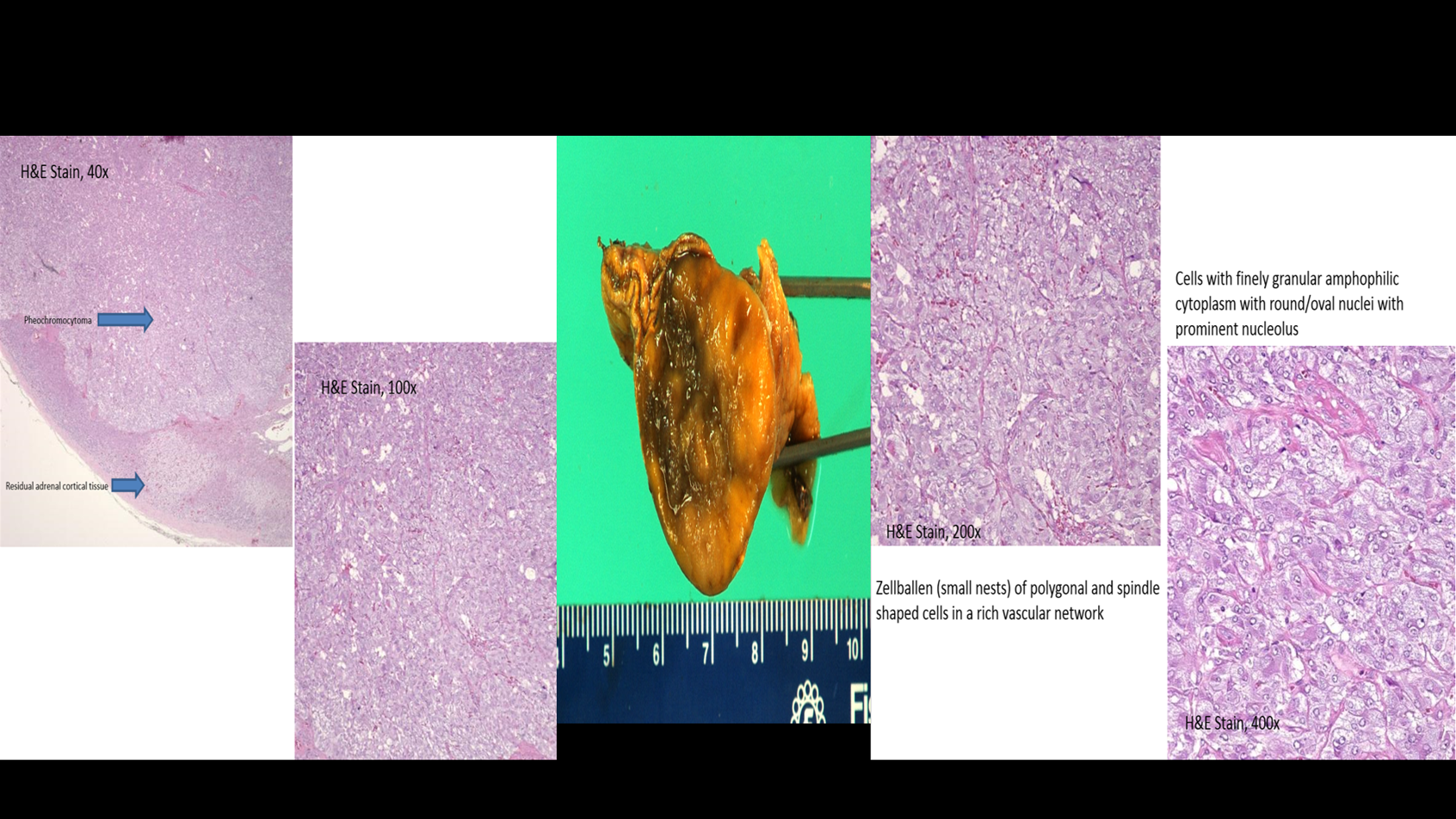
Based on the patient’s history, additional laboratory studies were collected which were significant for elevated plasma metanephrines and normetaneprhines. Adrenal CT scan revealed a 3.1 cm right adrenal mass with 67% enhancement washout. The patient was started on phenoxybenzamine and metoprolol therapy and later underwent successful surgical resection of the mass.
Discussion: Pheochromocytoma is a rare adrenal tumor affecting 1 to 2 patients per million population. Patients may not always have the classic triad of symptoms. As our case demonstrates, patients may present with adrenergic crisis after induction of anesthesia and after administration of certain antihistamine medications. When pheochoromytoma is suspected, initial testing should include urine or plasma metanephrine levels. Elevated levels should prompt imaging with CT or MRI to localize the tumor.
Conclusions: Pheochromocytoma is a rare treatable cause of hypertension. It can be triggered by certain “innocent medications”. A high clinical suspicion along with focused, detailed history taking can lead to successful diagnosis. Patients awaiting surgical resection should be started on alpha blockade followed by beta blockade to prevent intraoperative adrenergic crisis. After surgical resection, the patient and their family should be carefully monitored and have biochemical and genetic testing to screen for recurrent disease and familial syndromes associated with pheochromocytoma such as Multiple Endocrine Neoplasia type 2, Neurofibromatosis type 1, and Von Hippel Lindau.