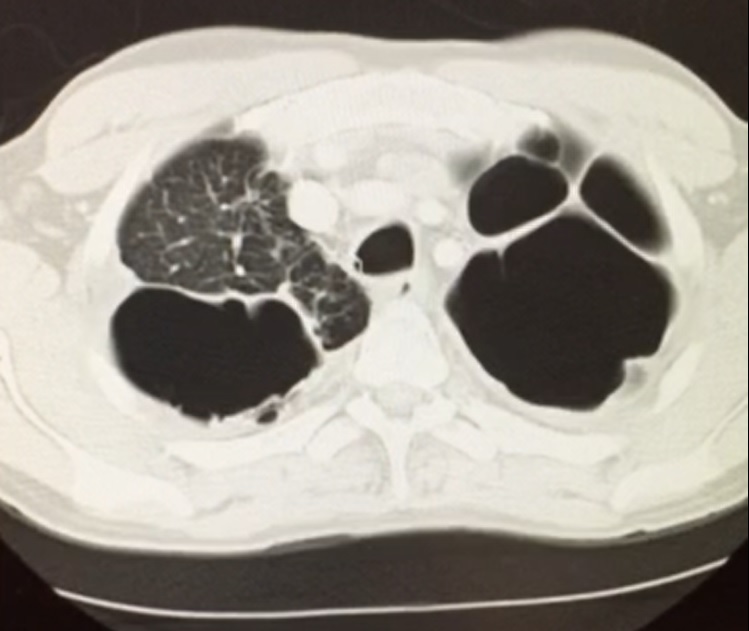
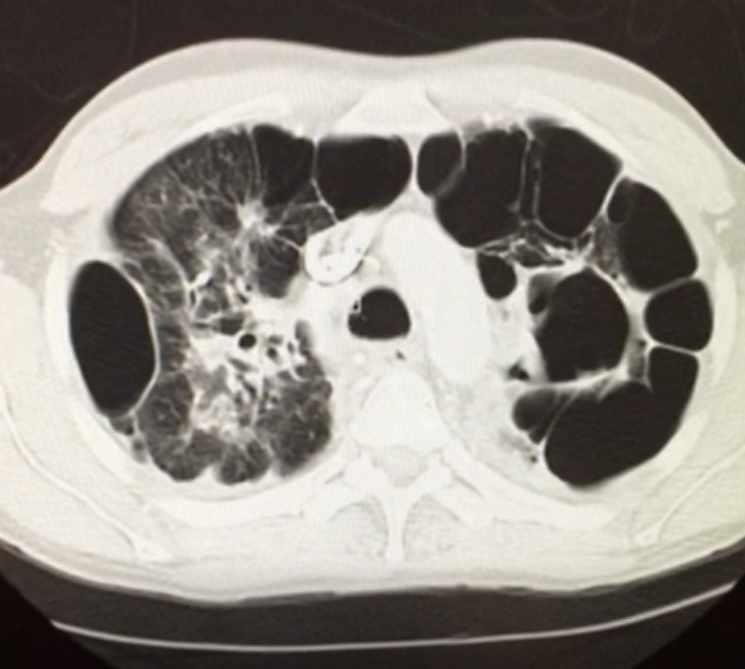
Case Presentation: A 38-year-old Guyanese man with a history of pulmonary tuberculosis 16 years ago, treated with a standard four-drug regimen, presented with an acute exacerbation of chronic cough which he had suffered for 12 years. Patient also endorsed a history of infertility. Chest CT showed significant parenchymal damage including multiple cavitations and bronchial thickening consistent with bronchiectasis (see Figures 1 and 2). Studies were notable for sputum cultures with multi-drug resistant pseudomonas with negative fungal studies, alpha-1 antitypsin levels within normal limits, three negative tuberculosis nucleic acid amplification tests, negative HIV and normal quantitative immunoglobulin testing, normal serum IgE levels, and nasal ciliary biopsy with no pathology seen. Genetic workup revealed a single mutated cystic fibrosis transmembrane conductance regulator gene (CFTR). Sweat chloride test returned positive. Treatment with inhaled antibiotics, hypertonic saline, and chest physical therapy provided significant symptomatic relief.
Discussion: The differential diagnosis for a young adult patient with bronchiectasis classically includes cystic fibrosis, damage secondary to prior pulmonary infection notably tuberculosis, primary ciliary dyskinesia (PCD), alpha-1 antitrypsin deficiency, immunodeficiency including HIV and immunoglobulin deficiency, allergic bronchopulmonary aspergillosis (ABPA), and underlying autoimmune disease. However, there are, in fact, a variety of CFTR-related disorders that should be a part of this differential. Heterozygous CFTR mutations result in a continuous phenotypic spectrum of pulmonary disease from asymptomatic patients to patients with diffuse bronchiectasis and positive sweat chloride testing. Patients with a single CFTR mutation with a 5T allele in the other CFTR gene can manifest with infertility from congenital absence of the vas deferens. Included in our differential was PCD, which can also present with chronic cough, infertility, and bronchiectasis on imaging. However, PCD features chronic rhinosinusitis and recurrent otitis media infections not seen in our patient. Screening can be performed via nasal nitric oxide testing with a confirmatory diagnosis made via ciliary or bronchial biopsy.
Conclusions: The differential for early-onset bronchiectasis includes a variety of CFTR-related disorders including CF homozygotes and CF heterozygotes, past pulmonary infection, PCD, alpha-1 antitrypsin deficiency, immunodeficiency states, ABPA, and autoimmune disease. Heterozygous CFTR mutations may present with a variable phenotype from limited asymptomatic disease to diffuse bronchiectasis, infertility, and a positive sweat chloride test.