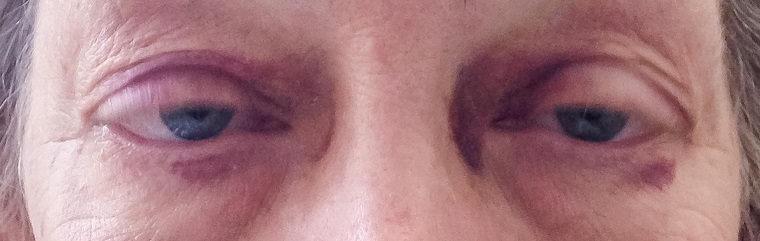
Case Presentation: A 65 year old female recently discharged from an outside hospital with new onset heart failure presented with 24 hours of worsening dyspnea associated with dizziness. Previous cardiac work up included an echocardiogram showing normal ejection fraction and normal cardiac catheterization. She was discharged on budesonide, carvedilol, losartan, and rosuvastatin. On presentation, she was afebrile, hypotensive and tachycardic, with normal oxygen saturation on room air. Physical exam was significant for periorbital purpura, bibasilar crackles and lower extremity edema. Labs revealed WBC 7.1 x 10e3, hemoglobin 16.6 g/dL, hematocrit 51%, platelets 397 x 10e3, troponin 0.828 ng/mL, N-terminal proBNP 12698 PG/ML, albumin 1.7 g/dL, urinalysis showed protein 500 mg/dL. Electrocardiogram showed low voltage and Q waves in the anterior, septal and inferior leads. Repeat transthoracic echocardiogram demonstrated severe concentric left ventricular hypertrophy, ejection fraction of 46-50%, mildly reduced left ventricular systolic function, transmitral spectral Doppler flow pattern suggestive of restrictive filling, mild right ventricular hypertrophy, and moderate pericardial effusion without tamponade. A cardiac MRI showed diffuse thickening of the ventricles with markedly abnormal gadolinium contrast dynamics and diffusely delayed myocardial gadolinium uptake indicative of an infiltrative process. An endomyocardial biopsy stained with Congo red demonstrated apple green birefringence under polarized light consistent with amyloidosis. Bone marrow biopsy was consistent with multiple myeloma. Treatment for the patient’s heart failure consisted of diuresis. Chemotherapy was started. The patient then developed sepsis from the bone marrow biopsy and subsequently passed away.
Discussion: Systemic amyloidosis is a multisystem disease caused by the extracellular deposition of various misfolded precursor proteins. Primary AL amyloidosis is a rare disorder caused by the extracellular deposition of monoclonal immunoglobulin light chains and is associated with plasma cell dyscrasias. Cardiac involvement occurs in about 50% of cases. A high suspicion is needed to identify the constellation of symptoms and signs that point towards amyloidosis. Common findings include fluid overload, pericardial effusion, proteinuria, diffuse low voltage in EKG, concentric ventricular thickening with normal ejection fraction on TTE. Definitive diagnosis is made with endomyocardial biopsy. Correct diagnosis of causative amyloid deposition, done by additional testing, is necessary given different treatments and different prognoses. Mainstay of heart failure treatment is supportive only and includes diuretics, as no other heart failure regimens are appropriate for cardiac amyloidosis. Periorbital purpura, with biopsy proven amyloid, is pathognomonic for AL amyloidosis. Despite treatment options with systemic chemotherapy, prognosis for AL amyloidosis is poor. Based on the Mayo Stage prognostic model, those not undergoing HSCT and those with elevated troponin levels and NT-proBNP levels have a median survival of four months and a five year survival rate of 8%.
Conclusions: Infiltrative cardiomyopathy is an uncommon cause of HFpEF, but should always be considered in new onset HF. Periorbital purpura, in the setting of HFpEF, is virtually pathognomonic for AL. Despite treatment options, prognosis for AL is poor.