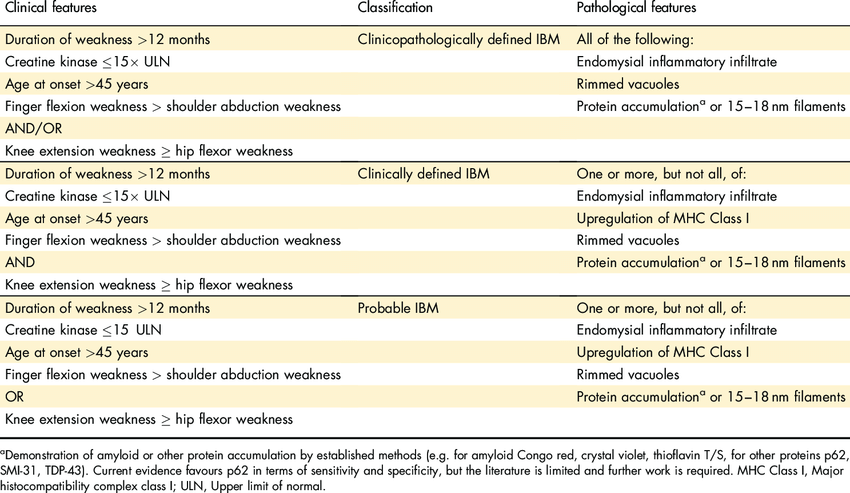
Case Presentation: A 74-year-old female patient presented to the clinic with a decade-long history of severe joint pain in her hands, escalating weakness in her arms and legs, difficulty gripping or holding objects, dysphagia, impacting daily activities. Past medical history includes sicca complex and fibromyalgia, with a family history of her sister having rheumatoid arthritis. Physical examination revealed weakness in the intrinsic muscles of the hands preventing the patient from making a fist, mild shoulder girdle weakness, proximal muscle weakness of thigh girdle, and tenderness of the glenohumeral joints. Lab: HLA–B27, ANA, RF negative, ESR 7, CRP < 0.3, CCP antibodies 15, aldolase 6.3, CK-MM 94, CK-MB 4, CK-MM 94, CK153, CK-BB 0, Serum myoglobin 246, CKI 327. Vectra score was moderate at 30. A muscle biopsy of the right vastus lateralis muscle displayed inflammatory infiltrates, foci of endomysial collections of T lymphocytes, scattered macrophages, increased connective tissue within the perimysium and endomysium. The patient's diagnosis of sporadic inclusion body myositis (sIBM) is confirmed as per the mandatory features, clinical features, and one pathologic feature (European neuromuscular center 2011 IBM diagnostic criteria).Steroid treatment yielded no response, although slight improvement was noted with Plaquenil, gabapentin and tramadol. The patient initially refused physical therapy and the use of a walker. However, after establishing a reliable rapport with the patient and her family, she agreed to utilize a walker and partake in physical therapy, committing to follow-up with a speech therapist.
Discussion: Sporadic Inclusion body myositis (sIBM), a rare inflammatory muscle disorder, is characterized by a reduced life expectancy and notably becomes the predominant myositis/inflammatory myopathy in individuals over the age of 50, with a prevalence of 18 per 100,000 people. It manifests as slow and progressive muscle weakness and atrophy, affecting various muscle groups. Dysphagia further compounds the morbidity. The average diagnostic delay is a significant 28 months. A comprehensive understanding of the patient’s medical history, coupled with a thorough physical examination, is crucial in reducing diagnostic delays for sIBM. Although the ENMC 2011 IBM diagnostic criteria exhibit impressive specificity (>99%), they do have limited sensitivity (57%). Regrettably, there is no cure, making treatment a challenging endeavor.The pathophysiology of sIBM remains incompletely understood but appears to involve the convergence of both inflammatory and degenerative pathways which is supported by the presence of highly differentiated CD8+KLRG1+ T cells, Anti-MUP 44 antibody targeting cN1A, and Beta amyloid in muscle fiber. Recent findings highlighting CD8+KLRG1+ T cells as contributors to the pathophysiology and potential treatment targets open new avenues for therapeutic exploration.
Conclusions: Unfortunately, traditional immunosuppressive treatments have proven ineffective in the treatment of sIBM. Dysphagia and impaired mobility significantly diminish the patient’s quality of life and lead to disability. Initiating physical therapy, rehabilitation, utilizing assistive devices, and securing strong family support significantly enhances muscle strength and overall quality of life, and should commence as early as possible.