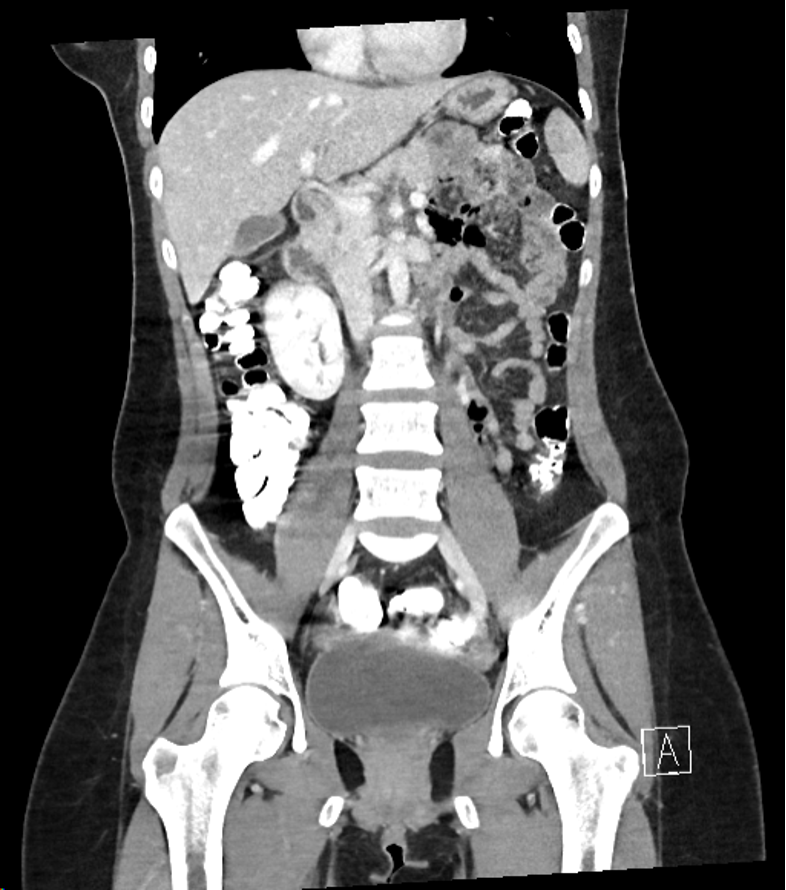
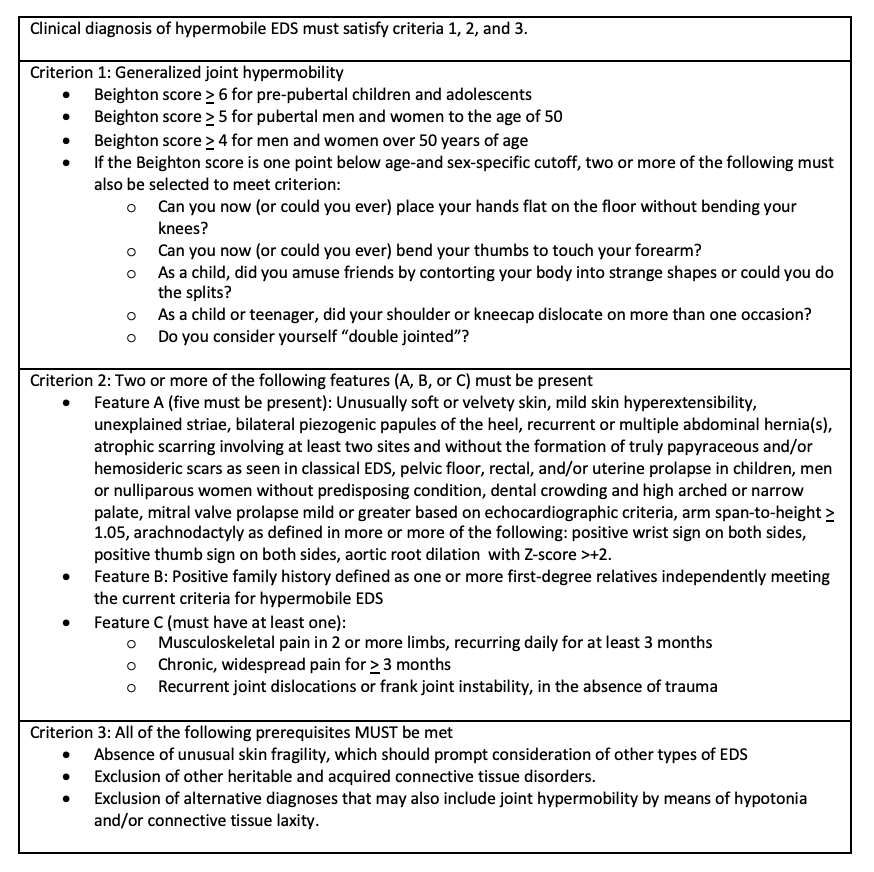
Case Presentation: A 27-year-old woman with a past medical history of recurrent bilateral shoulder dislocations presented with three months of progressive constipation, early satiety, nausea, vomiting, and an 18 kg unintentional weight loss. Prior to this admission, she had seen orthopedic surgery due to her recurrent, spontaneous bilateral shoulder dislocations and was noted to have hypermobile joints. She had no known family history of genetic or autoimmune disorders but reported that her brother has “tendon and ligament issues.” Labs were significant for hypokalemia (3.2 mmol/L), hypophosphatemia (2.1 mg/dL), anion gap of 17 mmol/L, and an elevated beta-hydroxybutyrate of 3.54 mmol/L. ECG revealed sinus tachycardia and CT abdomen/pelvis (figure 1) showed no acute abnormalities, but she had retained contrast from an upper gastrointestinal (GI) series that was obtained as an outpatient three week prior to admission. Based on evidence of prolonged retained contrast and clinical symptoms, she was diagnosed with GI dysmotility and started on prucalopride with improvement in her GI symptoms. After initial volume resuscitation and treatment of her starvation ketosis, she continued to have palpitations with standing, and vital signs were notable for an increased heart rate of 67 to 129 beats/min from supine to standing without any changes in blood pressure. She was diagnosed with postural orthostatic tachycardia syndrome and started on compression stockings and fludrocortisone. Given her constellation of findings, a unifying diagnosis of hypermobile Ehlers-Danlos syndrome was made as this can be associated with autonomic dysfunction. She met additional diagnostic criteria with hypermobile joints by Beighton score, velvety skin, skin hyperextensibility, unexplained striae, pelvic floor relaxation, and possible positive family history through her brother. She was managed with supportive care and referred to an EDS center for follow up.
Discussion: Ehlers-Danlos syndromes are a diverse group of hereditary connective tissue disorders that are characterized by joint hypermobility, skin hyperextensibility, and some variants can cause cardiac, vascular, ophthalmic, and dental issues (1). The overall prevalence of EDS is between 1 in 3,500 to 1 in 5,000 people (1). The Ehlers-Danlos syndromes are classified into 13 types, but the hypermobile subtype is the most common (2). Genetic variants have been identified in all types except for the hypermobile subtype (2). While there is no cure for EDS, diagnosis is important for symptom management. As with our patient, hypermobile EDS can be associated with GI dysmotility and autonomic dysfunction (1,2). In severe cases, laxatives and anti-emetics may not be adequate to restore bowel function leading to prolonged hospitalizations. Those patients with features of EDS and postural orthostatic tachycardia syndrome may benefit from earlier trials of alternative pro-kinetic agents (3).
Conclusions: Ehlers Danlos syndromes represents a heterogenous group of connective tissue disorders that are often not considered in the differential diagnosis of an inpatient setting. Increased awareness of clinical manifestations and diagnostic criteria is needed to prevent delays in timely care.