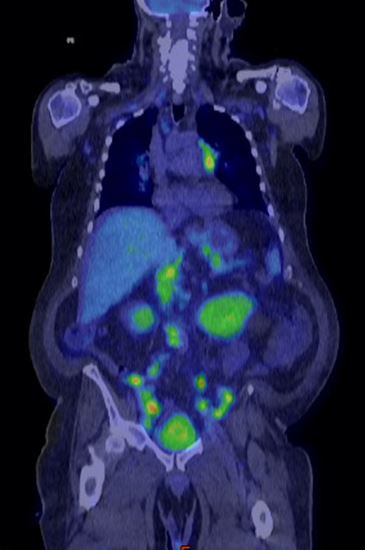
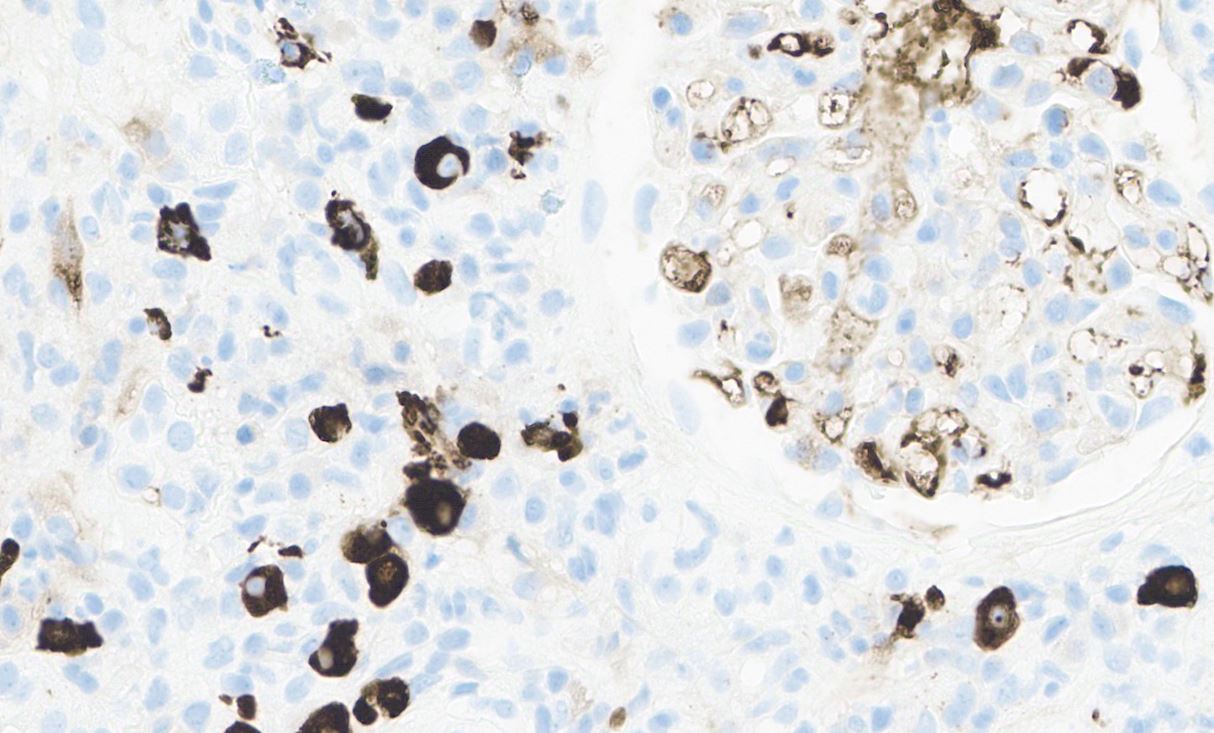
Case Presentation: A 68-year-old male with history of IgG4-related autoimmune pancreatitis presented to the Emergency Department with a one-month history of generalized weakness and a 25 lb unintentional weight loss over the past three months. Labs were notable for elevated lactate (3.4 mmol/L), new thrombocytopenia (platelets 29 ×10^9/L), and a stage II acute kidney injury (AKI) with creatinine 2.4 mg/dL compared to baseline 1.1 mg/dL. Computed tomography (CT) of the abdomen and pelvis showed widespread abdominopelvic lymphadenopathy and splenomegaly (spleen length: 16.5 cm), concerning for hematologic malignancy. Hematologic work-up revealed a biclonal gammopathy with IgG kappa and IgG lambda M spikes (0.7 mg/dL and 0.8 mg/dL respectively). Fluorodeoxyglucose (FDG) position emission tomography (PET)/CT showed intensely FDG-avid lymphadenopathy above and below the diaphragm (Figure 1), compatible with a lymphoproliferative disorder. There was intense FDG uptake associated with opacities in the bilateral lung bases, concerning for involvement of “lymphoma” in the lungs. Lymph node biopsy was negative for hematologic malignancy; 10% of the plasma cells in the lymph node expressed IgG4. Bone marrow biopsy showed a minute (< 1%) population of monoclonal B cells, consistent with MGUS. Kidney biopsy (Figure 2) showed IgG4-rich plasma cells with storiform fibrosis indicative of IgG4-related tubulointerstitial nephritis. Broad infectious work-up including Karius testing was negative. The patient was initiated on rituximab and steroids for widespread relapse of IgG4-related disease (IgG4-RD) with favorable clinical response including rapid improvement of his AKI and near resolution of lymphadenopathy on interval PET/CT three months after hospital discharge.
Discussion: IgG4-RD is an autoimmune fibroinflammatory disorder in which IgG4-secreting plasma cells invade various tissues, contributing to inflammation and scarring. Any organ system can be impacted by IgG4-RD, though salivary glands, lacrimal glands, and the hepatopancreaticobiliary system are most commonly involved. Correctly identifying IgG4-RD requires a high index of clinical suspicion. It is important to communicate this clinical suspicion to the reading pathologist to ensure immunohistochemistry for IgG4 is performed on the biopsy tissue. In our patient, diagnosis was delayed due to anchoring bias after initial CT A/P and subsequent PET/CT were read as highly concerning for lymphoma. Widespread relapse of IgG4-RD was not considered in the initial differential as the patient had limited health literacy, and his history of IgG4-related pancreatitis was not clearly documented in the Electronic Health Record (EHR). The rate of IgG4-RD recurrence is as high as 36% in the three years after initial diagnosis (1), and it is crucial for clinicians who will care for impacted patients in the future to be aware of this clinical history. Treatment of relapsed IgG4-RD involves combination therapy with glucocorticoids and other immunosuppressants including azathioprine, mycophenolate mofetil, or rituximab. Rituximab most effectively reduces IgG4-RD relapse rates compared to glucocorticoids and other immunosuppressive agents (2).
Conclusions: IgG4-RD should remain in the differential diagnosis for patients with widespread FDG-avid lymphadenopathy. The relapse rate is nearly 40% (1), and this case highlights the importance of distinctly documenting a history IgG4-RD in the EHR to guide future clinicians.