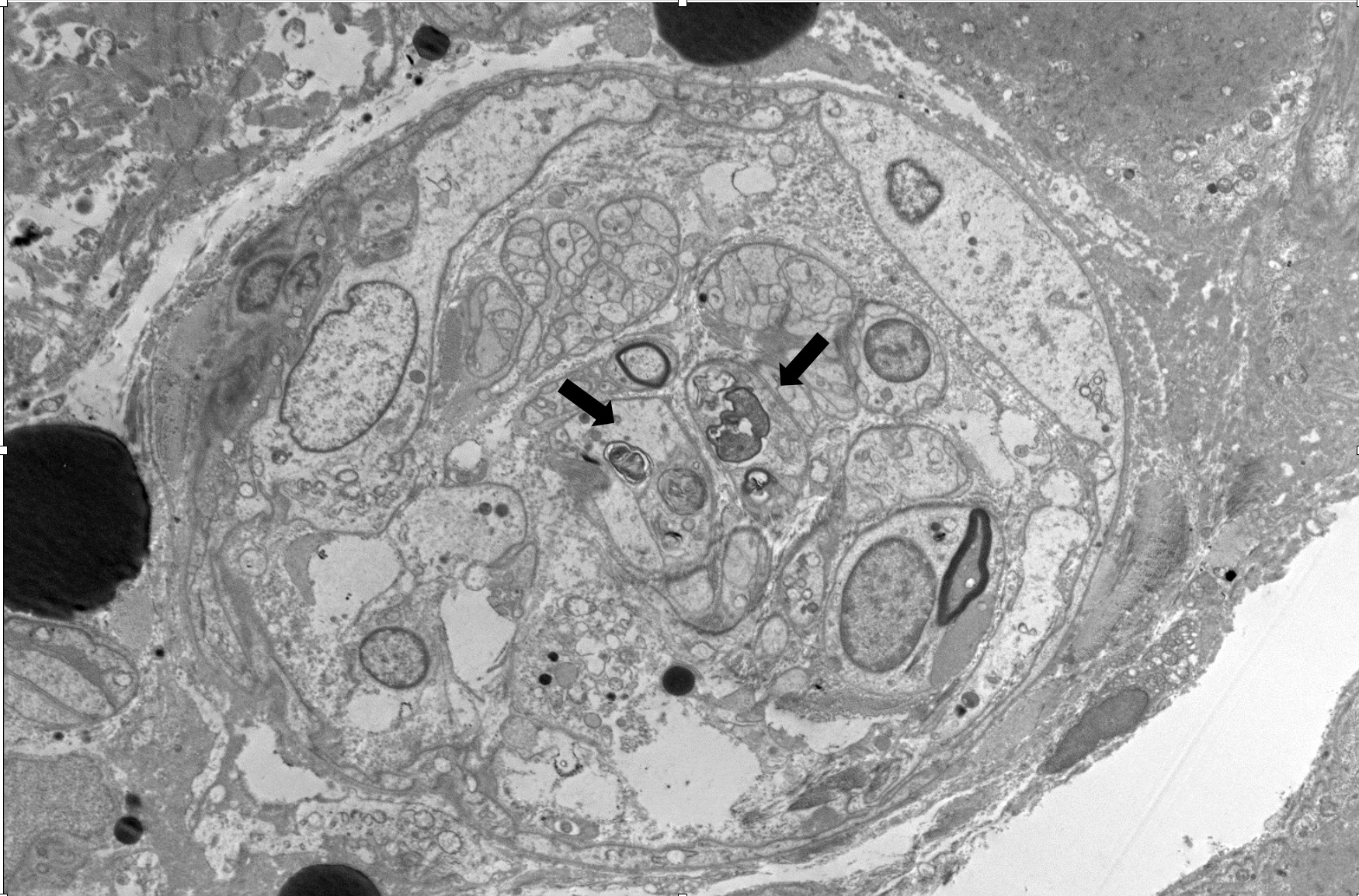
Case Presentation: 22-year-old female presented with worsening lower abdominal pain, diarrhea, and ascending muscle weakness. A lumbar puncture revealed a normal cell count and protein levels, with slightly elevated Campylobacter Jejuni antibody levels at 0.93. The patient was treated with a five-day course of IVIG for presumed Guillain-Barré Syndrome (GBS), resulting in slight improvement in muscle strength following rehabilitation; however, she remained wheelchair-bound. Three months later, the patient presented with recurrent episodes of diarrhea, severe abdominal pain, insomnia, and visual hallucinations. The patient was found to have flaccid quadriparesis with no antigravity strength and areflexia with elevated CK 7949U/L. Imaging studies, including MRI of the head and cervical spine, were unremarkable, while repeat lumbar puncture revealed isolated slightly elevated protein levels of 47mg/dL. Despite another five-day course of IVIG treatment, there was no significant improvement in motor function. Muscle biopsy showed muscle atrophy, demyelination and denervation pattern, ruling out myositis. Extensive diagnostic tests for various autoimmune and metabolic conditions, including serum heavy metal, ANCA, AchR antibody, SSA / SSB, RNP antibody, scleroderma antibody, centromere antibody, complement, and TSH, were all negative. The patient’s clinical course was complicated by severe generalized pain, persistent tachycardia, and urine discoloration resembling a wine color. Laboratory investigations revealed elevated levels of plasma PBG 99 nmol/mL (reference value < 0.5nmol/mL), spot urine PBG 96 µmol/mL (reference value < 0.22 µmol/mL), serum ALA 0.8 nmol/mL (reference value < 0.5 nmol/mL), along with a positive family history, confirming the diagnosis of acute intermittent porphyria. Hematology was consulted, and the patient was initiated on hematin at a dosage of 3 mg/kg/day for four days, followed by 4 mg/kg/day for 56 doses until serum PBG and ALA levels normalized. The patient then received a single dose of Givosiran. Gradual improvement was observed, enabling the patient to stand and walk with assistance. However, following the discontinuation of hematin, weekly spot urine PBG levels trended upward, hematin therapy was restarted.
Discussion: Acute intermittent porphyria (AIP) is a rare metabolic disorder characterized by a deficiency of the enzyme porphobilinogen deaminase, leading to the accumulation of porphyrin precursors, particularly porphobilinogen (PBG) and δ-aminolevulinic acid (ALA). Porphyria patients are rarely accurately diagnosed during the early stages of the illness.This case highlights the importance of considering AIP in the differential diagnosis of patients presenting with unexplained abdominal pain and neuropsychiatric manifestations, as early recognition and treatment are crucial in preventing life-threatening complications associated with acute porphyric attacks. Additionally, it demonstrates the importance for the clinician to be cognizant of the risk of anchoring bias when a patient has previously been given a potentially confounding diagnosis, such as GBS in our case. A thorough, accurate family history when considering alternative diagnosis is also important.
Conclusions: This case highlights the challenges in diagnosing AIP due to its nonspecific clinical presentation, emphasizing the significance of a high index of suspicion and timely biochemical testing to achieve a prompt diagnosis and optimize patient outcomes.