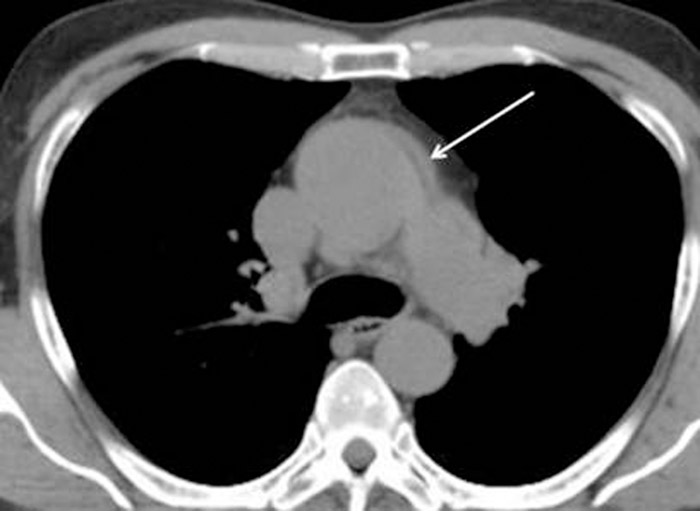
Case Presentation:
A 68 year‐old man with a past medical history of autosomal dominant polycystic kidney disease (ADPKD) and diverticulosis presented with a 1‐day history of severe chest pain radiating to the back with associated diaphoresis and dizziness. Vital signs were within normal limits; blood pressure measured 102/63 mmHg with no significant difference between left and right arms. Physical exam revealed no abnormalities. Chest radiograph demonstrated a widened mediastinum and tortuous aorta. A subsequent chest CT scan (without contrast due to creatinine of 2.1 mg/dL) showed a dilated aortic root (4.8cm) with an intramural hematoma of the ascending aorta and aortic arch, consistent with Stanford type A aortic dissection (Figure). Bedside transesophageal echocardiogram (TEE) confirmed severe aortic dilatation, type A aortic dissection (with a false lumen extending from the aortic root to the arch) and severe aortic regurgitation. The patient was emergently taken for surgical repair of the dissection, placement of a Hemashield tube graft, and aortic valve repair. Despite an intraoperative aortic rupture, the patient was successfully treated and recovered completely with discharge 10 days later.
Discussion:
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most commonly inherited diseases, with complications including end‐stage renal disease (ESRD) and extra‐renal manifestations such as intracranial aneurysms, hepatic and pancreatic cysts, diverticular disease, cardiac valve disease, and various other vascular abnormalities. Traditionally, ADPKD associated thoracic aortic aneurysm and dissection are believed to be a consequence of ADPKD related hypertension and ESRD requiring renal replacement therapy. However, as demonstrated by our case, these serious complications can also occur even in the absence of hypertension and ESRD due to PKD‐1 and PKD‐2 mutations leading to loss of vascular structural integrity. It is critical for hospitalists to recognize the potential life‐threatening complications of ADPKD, particularly in patients presenting with headache or chest pain. Even when suspicion for aortic dissection is relatively low, ADPKD patients should be rapidly evaluated with either chest CT angiography or TEE. This case also highlights the importance of recording a detailed family history, which may help to identify previously undiagnosed ADPKD as well as familial thoracic aortic complications in ADPKD (which has been previously reported in the literature). Large multicenter cohort trials are warranted to fully assess the incidence of ADPKD associated thoracic aortic pathology as well as genetic penetrance and expressivity.
Conclusions:
Thoracic aortic dissection should be considered in patients with ADPKD presenting with chest pain, regardless of prior history of hypertension or ESRD. If suspected, urgent imaging with CT angiography or TEE should be performed.