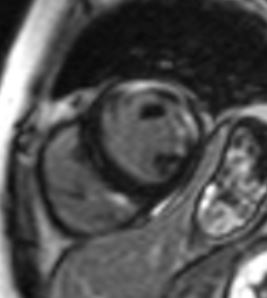
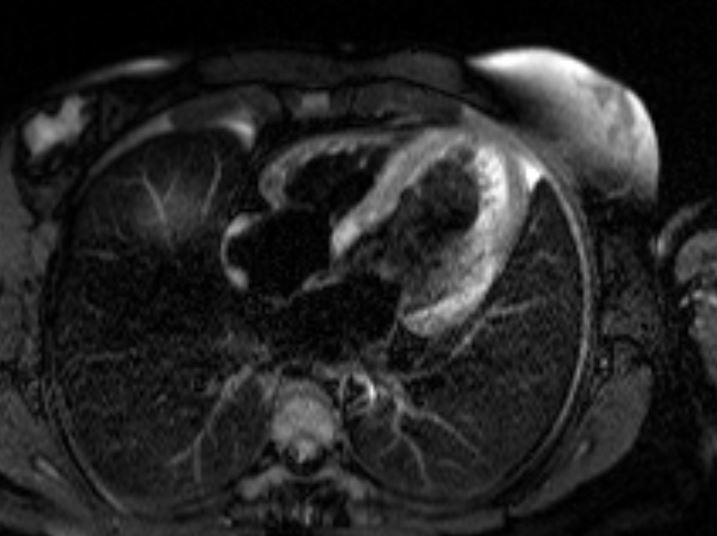
Case Presentation: A 32-year old woman with past medical history significant for myasthenia gravis (MG), polymyositis, and systemic lupus erythematosus (SLE) was admitted to the hospital for evaluation of non-sustained ventricular tachycardia (NSVT) discovered during 72-hour Holter monitoring. Patient had been experiencing palpitations with heart rate up to 200 beats per minute recorded on her smart watch, associated with substernal chest pressure and exertional dyspnea, for one month. Her medications included pyridostigmine, mycophenolate mofetil and prednisone; she was also receiving monthly plasmapheresis due to severe exacerbation of myasthenia symptoms 4 months prior. Labs on presentation were significant for troponin elevation at 10.6 ng/mL (normal: <0.04), BNP 462, hypogammaglobulinemia, hypocomplementemia, and normal ESR and CRP. CT Angiogram of the coronaries was negative for atherosclerosis. Hypokinesis of the inferior myocardial wall was noted on echocardiogram. Cardiac MRI showed reduced left and right ventricular systolic function, with a left ventricular ejection fraction (LVEF) of 41%. Multiple biventricular areas of myocardial enhancement were present, indicating an acute injury. Infectious workup, including a full viral panel, was negative. Review of skeletal muscle biopsy performed at an outside hospital showed giant cells, inconsistent with a diagnosis of polymyositis. Patient had previously 2 positive and 1 negative dsDNA antibody tests; repeat anti-dsDNA using a crithidia assay, which has the highest specificity for SLE, was negative. Hypocomplementemia and hypogammaglobulinemia were attributed to recent plasmapheresis. Further workup showed positive acetylcholine receptor binding, blocking, and modulating antibodies, as well as positive anti-titin and anti-striational muscle antibodies, all seen in MG. No thymoma was present on Chest CT. Treatment with intravenous high dose steroids and mycophenolate was initiated with resolution of ventricular arrhythmias. Plasmapheresis was switched to weekly intravenous immunoglobulin (IVIG) administration. Amiodarone was started for arrhythmia control. Follow-up echocardiogram 7 days after treatment initiation showed improved LVEF of 45-49%.
Discussion: Myocarditis is a very rare manifestation of MG; furthermore, its pathogenesis is not clearly understood. A handful of cases can be found in literature, majority of which involve patients with increased age, thymoma, and anti-striational antibodies, suggesting a possible autoimmune or paraneoplastic etiology. Voltage-gated potassium channel antibodies have been associated with a more fatal giant cell myocarditis in a subset of patients with MG. Here we report a case of myasthenia-related myocarditis in a young patient without thymoma, voltage-gated potassium channel antibodies, or underlying coronary artery disease. Positivity of acetylcholine receptor and anti-striational antibodies, in the absence of other insults to the myocardium, supports this diagnosis. Additionally, the presence of giant cells in the skeletal muscle specimen raises the possibility of an even rarer case of giant cell myocarditis.
Conclusions: Myasthenic myocarditis should be considered in patients with underlying MG presenting with non-ischemic cardiomyopathy or unexplained ventricular arrhythmias, even in the absence of a thymoma. Treatment with IVIG, high dose steroids, mycophenolate and anti-arrhythmic drugs have proven to be effective in preventing recurrences.