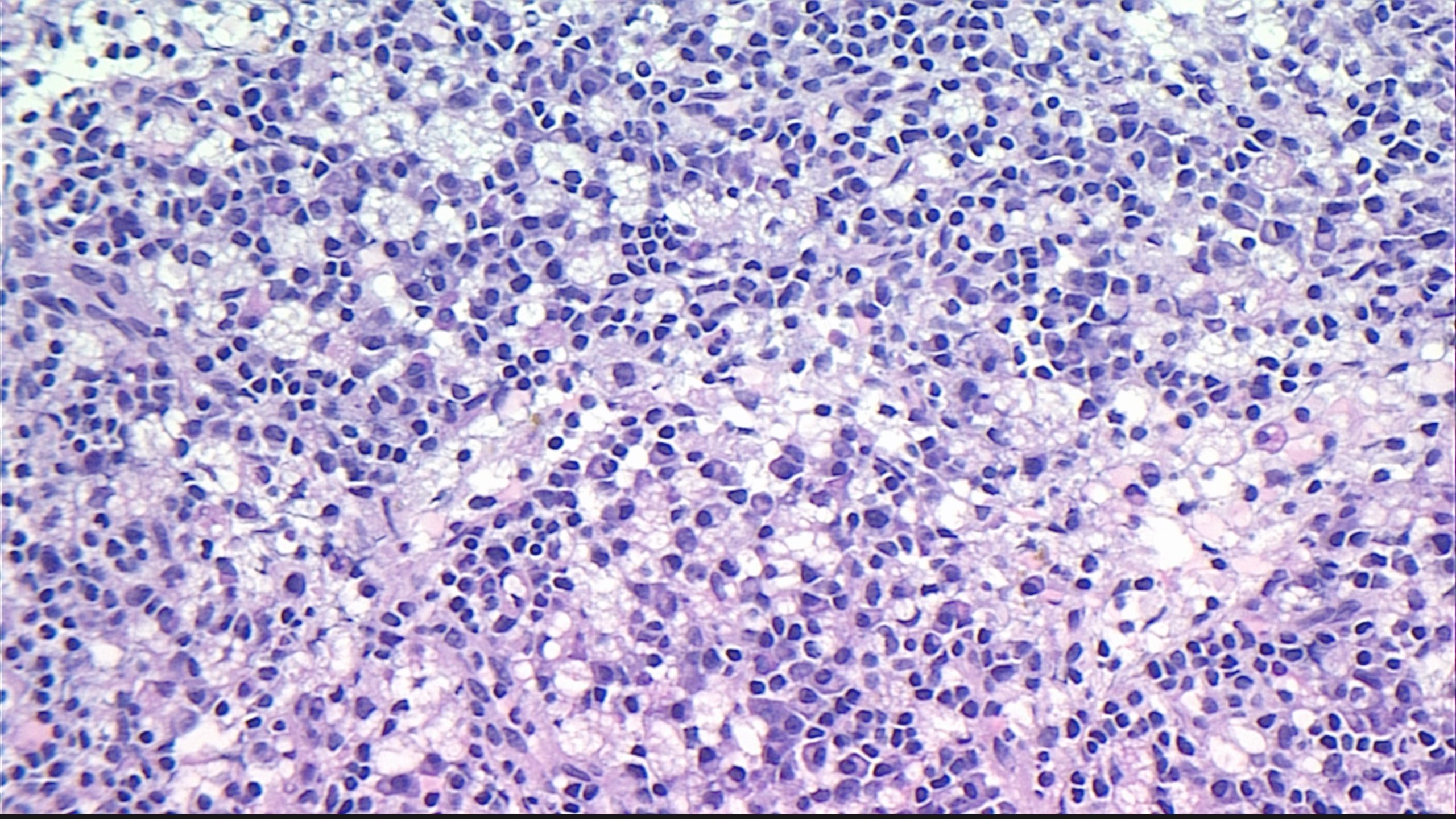
Case Presentation: A 49 year old female with past medical history of atrial fibrillation, cerebrovascular accident, and hypertension presented with abdominal pain and rectal bleeding. Patient reported 3 days of worsening, non-radiating L-sided abdominal pain. Review of systems was positive for one episode of fever to 38.3℃, nausea, anorexia, and blood-streaked diarrhea. The patient had been admitted for similar complaints 4 months prior, but symptoms improved with supportive measures. At the time, she had had a CT scan of her abdomen and pelvis which demonstrated diffuse nonspecific retroperitoneal and mesenteric lymphadenopathy, but no further workup was done at the time. Repeat CT of the abdomen and pelvis upon admission demonstrated stable lymphadenopathy. Patient underwent a broad workup, which was negative for typical and atypical infectious and autoimmune causes of lymphadenopathy. She also received a retroperitoneal core needle biopsy which demonstrated necrotizing lymphadenitis, consistent with Kikuchi-Fujimoto Disease. On H&E slides, characteristic proliferation of lymphoid cells with large areas of necrosis and foamy histiocytes were observed (Figure 1). Treatment was supportive, and symptoms improved by hospital day 2.
Discussion: Kikuchi-Fujimoto Disease (KFD) is a rare and self-limited lymphoproliferative disease. The most common presentation of KFD is fever, diarrhea and cervical lymphadenopathy. There have also been rare cases of reported retroperitoneal lymphadenopathy, as was seen in our patient. The etiology of KFD remains elusive. Viral etiologies have been described, however, the reproducibility of these results is limited. Notably, autoimmune etiologies have also been explored, primarily SLE, which has been reported to co-occur with KFD several times.Familiarity with this rare disease is important to hospitalist medicine given the “red-flag” symptoms associated with KFD, including lymphadenopathy, fevers, and night sweats. As such, a patient who presents with this disease is likely to be worked up thoroughly and invasively for malignancy, autoimmune and infectious etiologies. Even though KFD is benign and self-limited, diagnosis remains only possible through lymph node biopsy, demonstrating necrotizing lymphadenitis. It is also important for the hospitalist physician to be aware of the relapsing rate of KFD, which is as high as 11%. In a patient like ours, with recurrent nonspecific symptoms and previously documented and stable lymphadenopathy, it is reasonable to have KFD in the differential diagnosis. Additionally, KFD may co-occur with SLE, so it is important to send ANA and dsDNA markers on patients diagnosed with KFD. Some reports have called for monitoring of patients with KFD for the development of SLE, which is a consideration important to the hospitalist physician when establishing appropriate follow-up. Notably, the treatment of KFD is supportive. In the case of our patient, her symptoms resolved during the first few days of her admission; however as noted before, recurrence is not rare.
Conclusions: Kikuchi-Fujimoto Disease is a rare, benign and self-limited cause of lymphadenopathy. It should be considered in patients with nonspecific symptomatology and stable lymphadenopathy. Definitive diagnosis is through lymph node biopsy, which demonstrates necrotizing lymphadenitis. Treatment for patients is supportive, with spontaneous improvement in most cases. However, close attention should be paid to co-occurrence with SLE as well as recurrence.