Case Presentation: A 23-year-old male with HgB SS sickle cell disease and iron overload due to chronic transfusions presented from an outside hospital with sickle cell pain crisis complicated by a total bilirubin level of 30 U/L. At the time of admission, his pain crisis had resolved. Physical exam revealed scleral icterus and gallbladder ultrasound showed cholelithiasis with only trace ductal dilation. Hematology was consulted due to rising unmeasurable total bilirubin levels (>60 U/L). He had a red cell exchange without improvement in hyperbilirubinemia followed by plasma exchange transfusion (PLEX) twice with initial improvement followed by rebound hyperbilirubinemia. He was evaluated by the hepatology service due to concurrent rising INR for liver transplant evaluation. He was deemed not a candidate for liver transplant due to high risk of recurrence of hepatic dysfunction. The hyperbilirubinemia eventually stabilized a week after his last plasma exchange transfusion and ultimately trended down rapidly. He was discharged home with folic acid, desferiprone, and ursodiol and established care with outpatient hematology to begin monthly red cell exchanges. Follow-up labs showed further decrease in total bilirubin.
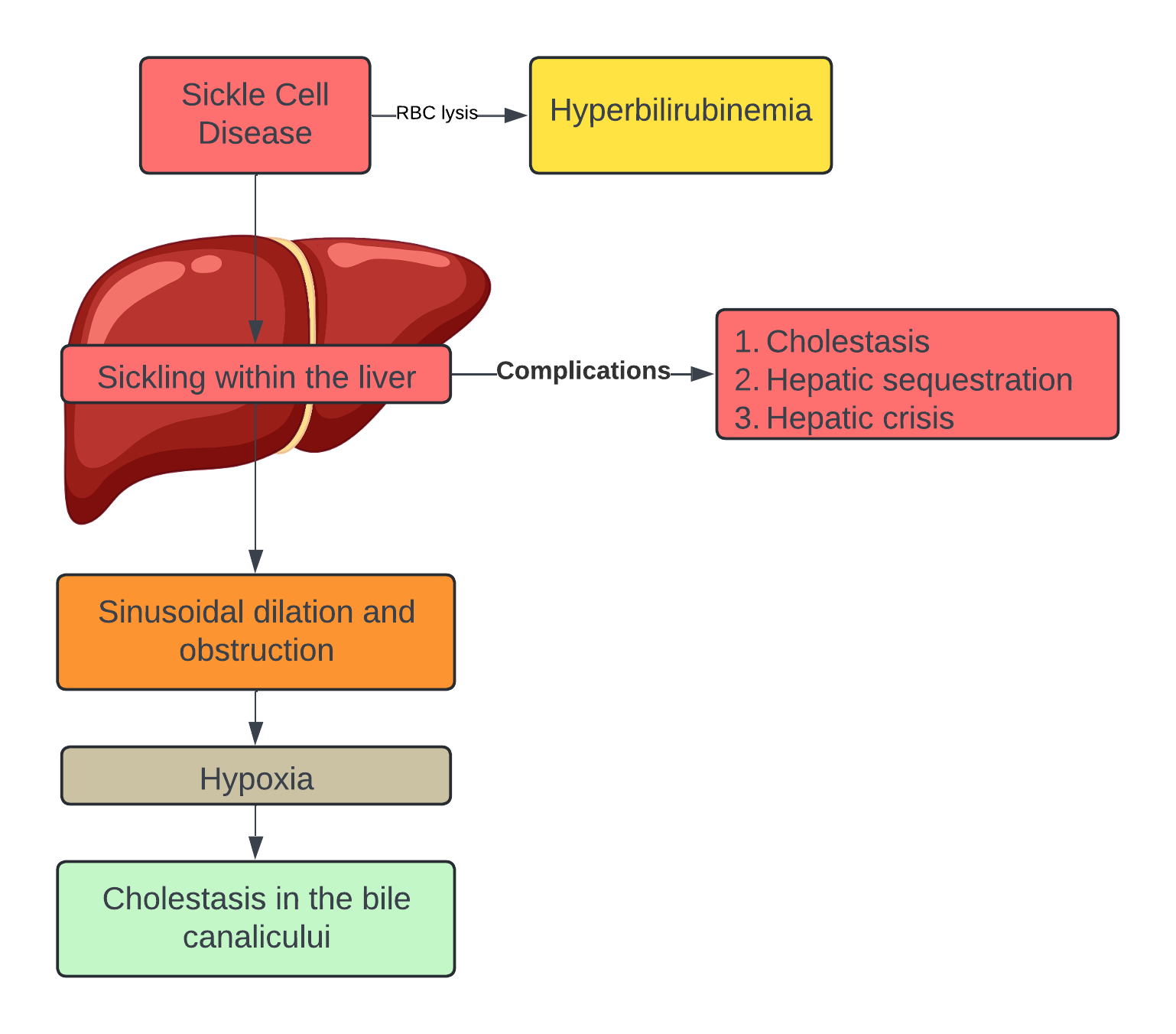
Discussion: Sickle cell pain crisis often leads to hyperbilirubinemia due to lysis of red blood cells though sickling within the liver can lead to further complications such as hepatic crisis, sequestration, or cholestasis. Sickling of the RBCs leads to sinusoidal dilation and obstruction leading to hypoxia of the hepatocytes with cholestasis in the bile canaliculi. It is important to recognize and screen rising transaminases, hyperbilirubinemia, and rising INR in sickle cell patients to assess acute intrahepatic cholestasis. Management of intrahepatic cholestasis starts with supportive measures though if these do not result in improvement in hepatic function, hematology should be consulted to discuss the need for PLEX and fresh frozen plasma to reverse coagulopathy of hepatic dysfunction. As in the case of our patient, if PLEX does not resolve cholestasis, hepatology should be consulted for liver transplant evaluation. Ursodiol can be used as well as it has shown some benefits.
Conclusions: Liver disease is common amongst patients with sickle cell disease and some will develop acute cholestasis. These patients should be screened early for elevated transaminases, hyperbilirubinemia, and elevated INR. If improvements are not achieved with supportive care, hematology should be consulted early to initiate PLEX. If there is still no improvement, then hepatology should be consulted for liver transplantation evaluation.