Case Presentation: A 57-year-old man with congestive heart failure and coronary artery disease presented with shortness of breath of two days duration. He denied fevers, chills, chest pain, or cough. Presenting vitals were significant for oxygen saturation of 80%, requiring. All other vitals were within normal limits. Physical exam was unremarkable. His labs were significant for thrombocytopenia to 7 K/uL, lactate dehydrogenase(LDH) 1696U/L and ferritin 14428ng/mL. Finally, he was found to be COVID-19 positive. Due to severe thrombocytopenia, peripheral smear was sent and was revealing for 4-5 schistocytes per high power field, raising suspicion for thrombotic thrombocytopenic purpura(TTP). PLASMIC score on admission was 6 (72% chance of ADAMTS13 deficiency). He underwent plasma exchange (PLEX) for presumed TTP. Thrombocytopenia was refractory to PLEX, requiring platelet transfusions every 2-3 days. PLEX was subsequently discontinued due to lack of clinical response and after ADAMTS-13 returned >10% at 15.2%. The patient was subsequently started on IV immunoglobulin(IVIG) for presumed immune thrombocytopenic purpura(ITP), also without platelet response. As thrombocytopenia was refractory to PLEX and IVIG, bone marrow biopsy was performed. Biopsy was revealing for hypercellular marrow with tri-lineage hematopoiesis and increased histiocytes with presence of hemophagocytosis. Further lab testing revealed elevated soluble IL-2 receptor at 3338pg/mL and triglycerides at 390mg/dL. CD56+/CD3- natural killer cell levels were decreased at 13cells/uL. Patient’s H-score was elevated at 165 and a diagnosis of hemophagocytic lymphohistiocytosis (HLH) was subsequently made. The patient received high dose steroids throughout his hospital course, however etoposide was deferred due to bacteremia. Patient ultimately died from multiorgan failure.
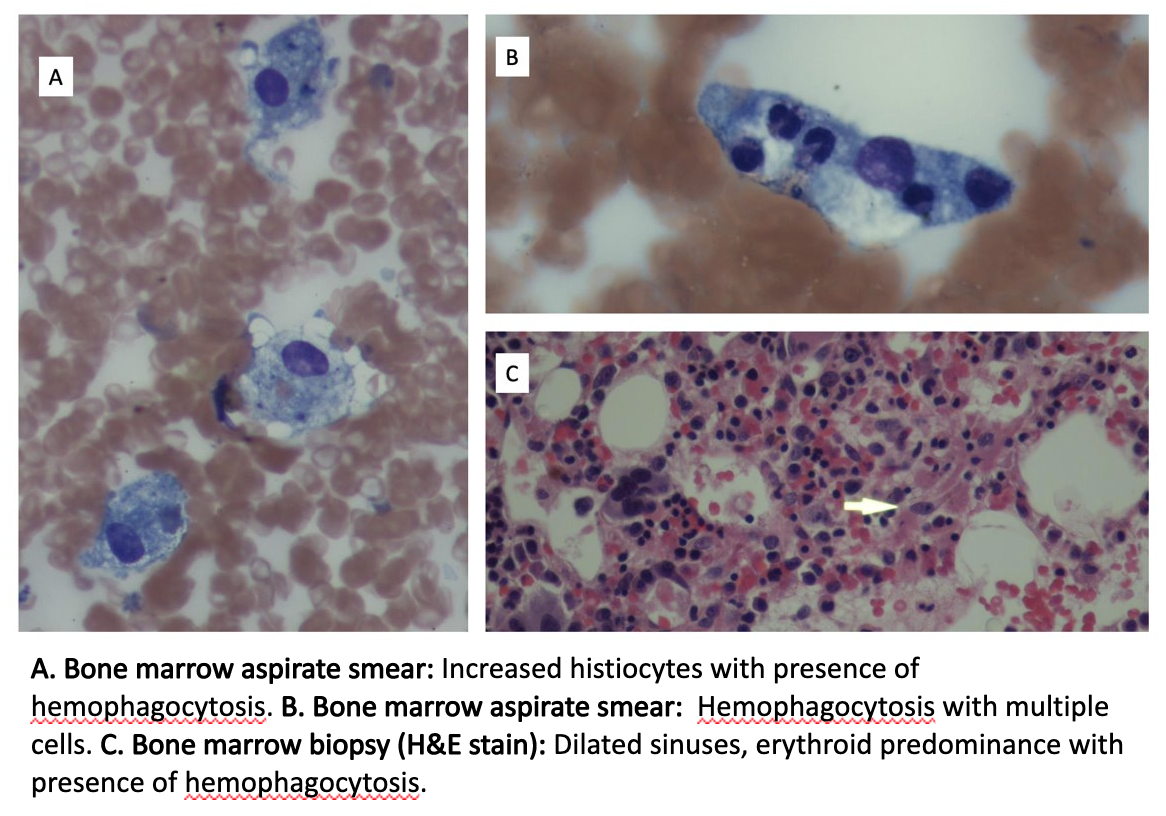
Discussion: Our patient was diagnosed with hemophagocytic lymphohistiocytosis (HLH) secondary to COVID-19. The diagnosis of secondary HLH can be difficult as COVID-19 is commonly associated with a multi-system inflammatory response that can mimic a typical HLH presentation. HLH from a hyperinflammatory state generally occurs from immune activation gone awry. This is a product of excessive cytokine release from activated macrophages that result in tissue damage and subsequent organ failure. The HLH-2004 study, reported common clinical findings in patients with suspected HLH. This includes fever, cytopenias, hypertriglyceridemia, splenomegaly, decreased natural killer cell activity, hemophagocytosis in biopsy, and increased ferritin and IL2 receptor levels. A diagnostic scoring system or H-score, was developed to help estimate probability of HLH. An H-score ≥ 250 signifies 99% probability of HLH and a score ≤ 90 signifies a < 1% probability of HLH. Our patient’s H-score was 165, conferring a 40-54% probability of HLH. Bone marrow aspirate revealed hemophagocytosis (Images A,B,C). Treatment usually consists of steroids and etoposide. Our patient had already received extensive steroids throughout his prolonged hospitalization and etoposide was ultimately deferred due to the patient’s florid multi-organ failure.
Conclusions: Many clinical manifestations of COVID-19 continue to remain unknown. Patients with refractory thrombocytopenia of unclear etiology should undergo COVID-19 testing. HLH or an HLH-like syndrome should be on the differential when evaluating thrombocytopenia in COVID patients.