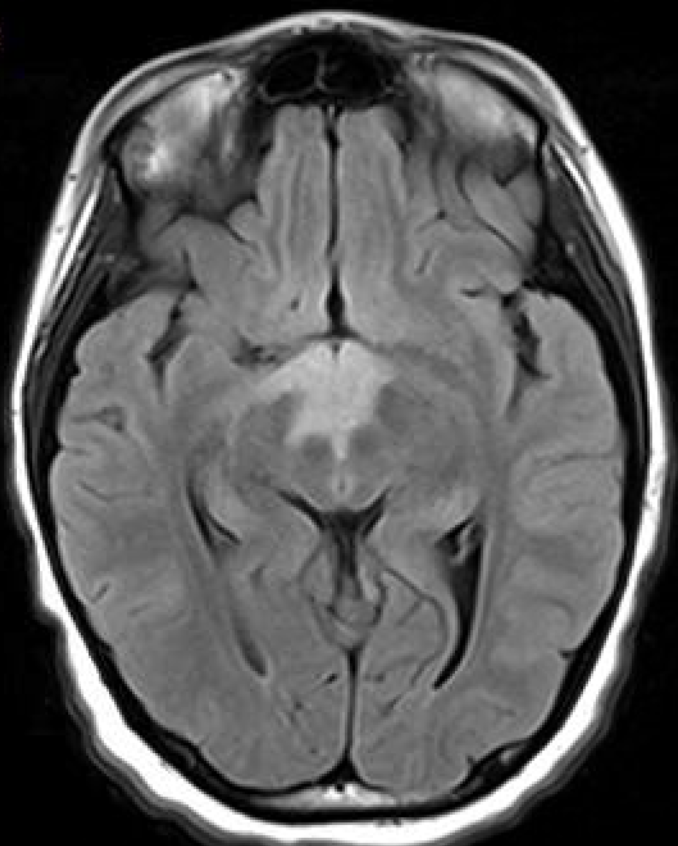
Case Presentation: 12-year-old girl presented with altered mental status, hypothermia, and bradycardia following one week of abdominal pain, progressive fatigue and flattened affect. Family denied ingestion, fever, cardiorespiratory symptoms, headaches or unusual exposures. Exam significant for Temperature 33.2 C, Heart Rate 42 bpm, sluggishly reactive pupils, disconjugate gaze, able to intermittently follow commands and localize to pain. Laboratory studies revealed hyponatremia, hypokalemia and negative infectious work-up. CSF analysis had a persistent lymphocytic pleocytosis and oligoclonal bands. MRI Brain showed a non-enhancing, ill-defined lesion involving the hypothalamus and majority of the diencephalon, eventually with scattered grey-matter lesions which showed hypercellularity on biopsy. NMO/AQP4 titer was positive in the CSF, but not the serum. She was diagnosed with probable Neuromyelitis optica (NMO) autoimmune encephalitis with predominantly hypothalamic involvement. IVIG had no effect. Steroid treatment provided minimal improvement. Rituximab resulted in near-resolution of symptoms.
Discussion: Autoimmune encephalitis (AE) is an increasingly recognized etiology of encephalitis. It remains challenging to diagnose due to its varied presentation and limited antibody testing. Common presentations include delirium, seizures, behavioral changes, autonomic instability, respiratory failure, coma, and sepsis. NMO typically presents with optic neuritis or transverse myelitis but can present as an AE and has been described in AE syndromes related to diencephalic and brainstem pathology, as in this case. About 65% of children with suspicion for NMO have positive AQP4-IgG in the serum but seronegativity should not exclude the diagnosis as AQP4-IgG may appear as late as 5 years after initial symptoms. Early recognition and treatment can lead to reduced morbidity and mortality. The absence of antibodies should not delay treatment if AE is suspected. Treatment includes immunomodulatory therapies. First line therapies include steroids, plasmapheresis, and IVIG with advancement to second-line therapies such as rituximab or cyclophosphamide if no clinical improvement is seen.
Conclusions: As in this case, AE should be suspected in patients with an acute or subacute onset of neurologic symptoms that correlate with signs of inflammation on CSF studies, histopathological tests, and/or imaging. Other etiologies including toxins, vasculitis, metabolic and infectious should be explored. Though there is often diffuse brain involvement, presentations may have specific symptoms related to the localization of the initial inflammatory process. Hypothalamic involvement is reported to be associated with NMO/AQP-4 antibody-mediated encephalitis, though this patient lacked other typical symptoms associated with neuromyelitis optica (NMO). A history of autoimmune conditions as well as a preceding infectious prodrome support the diagnosis. Immunomodulatory therapy should be initiated as soon as infectious etiologies have been excluded to minimize morbidity and mortality. Clinical response should guide therapy, though treatment may take days-to-weeks to take effect and overall improvement in AE is often slow.