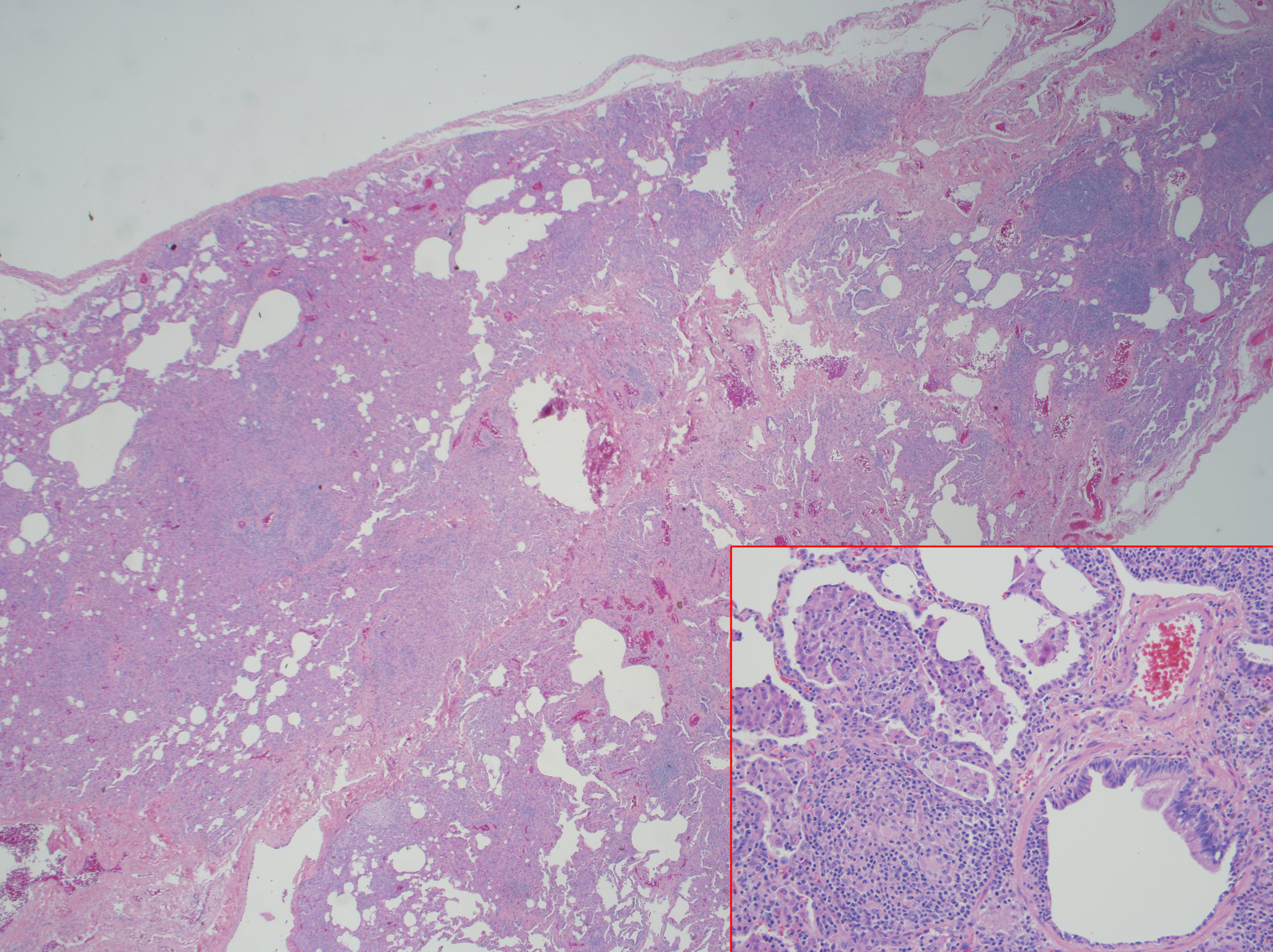
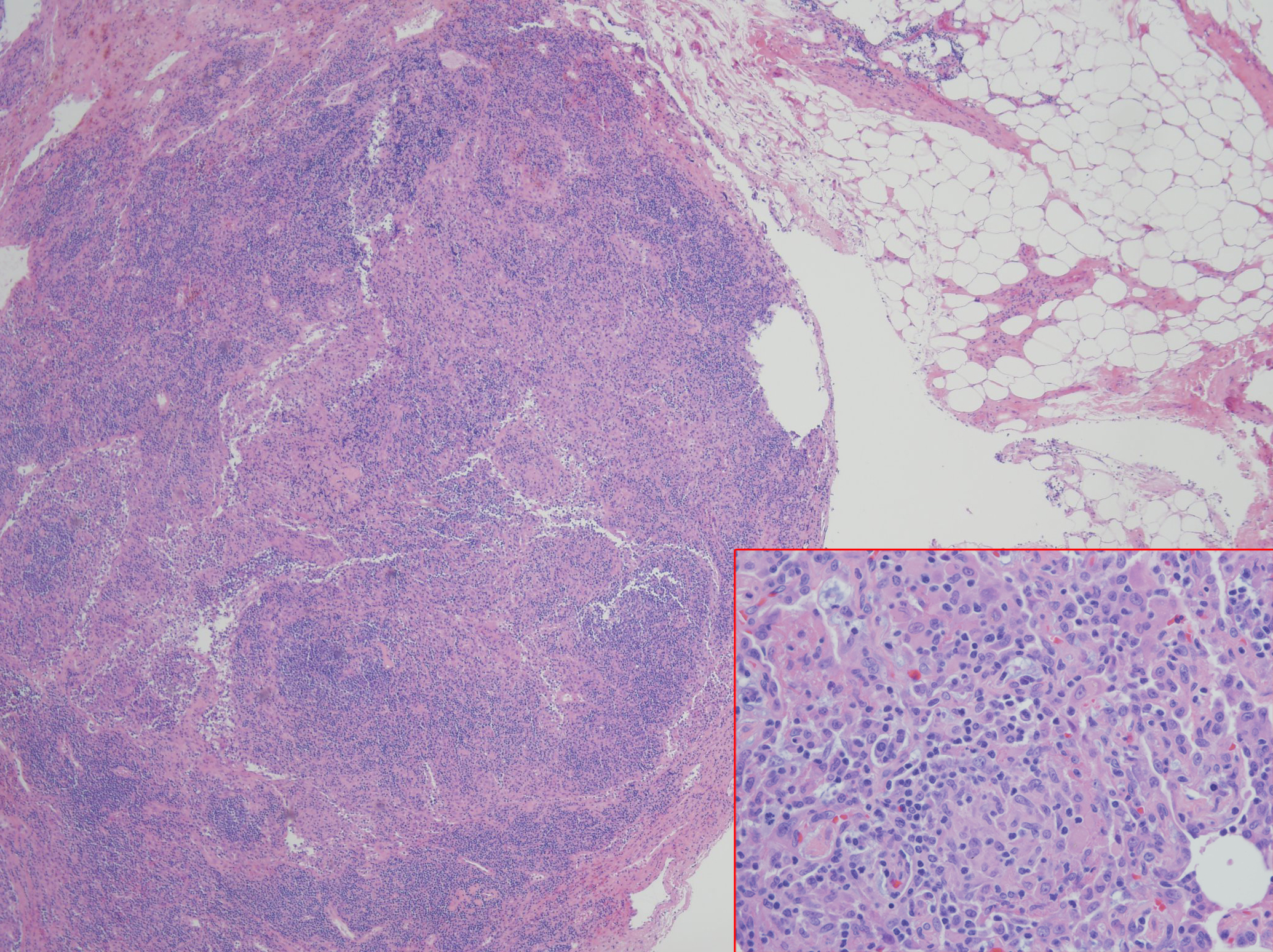
Case Presentation: A 34 year old male with a history of CVID subclass 2 and 4, with low IgG level, absent IgA, liver nodular regenerative hyperplasia with portal hypertension, and immune thrombocytopenic purpura status post splenectomy presented with a month of nonproductive cough, mild episodes of hemoptysis, and shortness of breath. Physical exam revealed peripheral capillary oxygen saturation of 89% at rest and 85% with ambulation, multiple petechiae on upper chest and bilateral shoulders, spider angiomata, clubbing, and cyanosis on all upper extremity digits. Laboratory studies were notable for eosinophilia with lymphocytosis, alkaline phostphatase 266 U/L, Total bilirubin 3.6 mg/dL, Direct bilirubin 1.3 mg/dL, Indirect bilirubin 2.3 mg/dL, BNP 160 pg/mL, Tryptase 19.6 µg/L, negative Influenza and ANCA. Chest x-ray showed small lung volumes. Chest CT angiogram showed no pulmonary emboli, mild right upper lobe tree-in-bud opacities; diffuse lymphadenopathy without changes from CT done at diagnosis of CVID. Pulmonary function test was normal. Transthoracic echocardiogram revealed an ejection fraction (EF) of 56-60% with an interatrial shunt and a mildly dilated left ventricle. Cardiac catheterization demonstrated a small patent foramen ovale (PFO) with right-to-left shunt, which was also demonstrated on V/Q scan. Bone marrow biopsy showed no evidence of malignancy. Bronchoscopy cultures revealed no infectious etiology. Given results above, hypoxemia was attributed to a combination of PFO and chronic lymphangitic changes with enlarging lymphadenopathy, unable to rule out lymphoma. Lymph node biopsy did not yield diagnostic result. Therefore, left video-assisted thoracoscopy with biopsy demonstrated non-necrotizing interstitial granulomas and follicular lumphoid hyperplasia. This finding was diagnostic for GLILD, a rare pulmonary disease that explained the patient’s hypoxemia. He improved significantly with rituximab followed by azathioprine, and intravenous immunoglobulin (IVIG).
Discussion: CVID is a lifelong primary immunodeficiency disorder characterized by impaired B-cell differentiation, low immunoglobulin levels, recurrent bacterial infections and malignant disorders involving lung, gastrointestinal tract, liver, and spleen. Approximately 20% of CVID is complicated by granulamatous-lymphocytic interstitial lung disease (GLILD), which is a diffuse lung parenchymal disease characterized by non-necrotizing granulomas, lymphoid interstitial pneumonia, and follicular bronchiolitis on histopathology. GLILD is associated with significant morbidity and mortality and its etiology is unknown. Common initial presentation includes dyspnea but may be asymptomatic. GLILD can be challenging to distinguish from sarcoidosis and lymphoma. Diagnosis requires a surgical lung biopsy. Optimal therapies that follow the diagnosis are unknown, although prednisone and IVIG are considered to be the first line therapy in GLILD, especially those with lymphoproliferative disease. Therapy with a combination of rituximab and azathioprine was successfully in some cases.
Conclusions: CVID is a primary immunodeficiency disorder characterized by low immunoglobulin levels. CVID patients can develop GLILD, the presence of which is associated with significant morbidity and mortality and should be intervened early.