Case Presentation:
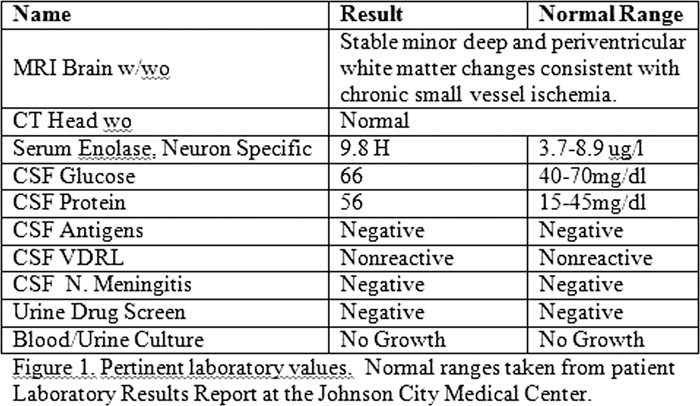
A 47 year old female was brought to our regional medical center by her son. She was aphasic, increasingly agitated, and markedly confused according to her son. Prior to arrival, she had been evaluated at two regional medical centers for stroke and diagnosed with acute bronchitis and conversion disorder. She saw Psychiatry outpatient, but continued to deteriorate. She stopped communicating one week prior to presentation, and started spitting out her pills. The son stated her first symptoms began abruptly including disorientation, confusion, slurred speech and weakness in the legs. Past medical history was significant for diabetes mellitus, chronic lumbago, tobacco abuse and chronic kidney disease. Psychiatric history included taking Celexa for assumed depression. Family history included schizophrenia, bipolar disorder and Alzheimer’s Disease. On exam, the patient was awake, moving spontaneously, disoriented, not following commands and withdrew to pain. She had myclonal jerks with an exaggerated startle response. Vitals were 100.2, 85, 20, 132/90. White blood cell count was 14.1, lactate 3.4, her radiographs were normal. Initial diagnoses were encephalopathy, sepsis, aphasia, and conversion disorder. Consults were done by Psychiatry and Neurology. The patient had extensive radiological, laboratory workup and lumbar puncture with pertinent findings shown in Figure 1. Her electroencephalography (EEG), had generalized periodic 1‐1.5 Hz sharp waves complexes, (normal range is 8‐13Hz). These findings were consistent with severe generalized cerebral dysfunction or diffuse encephalopathy. The presence of progressive neurodeneration, paucity of lab and radiographical findings, abnormal EEG and positive serum enolase indicated Sporadic Creutzfeldt‐Jakob disease (sCJD).
Discussion:
Creutzfeldt‐Jakob disease (CJD) is the most common type of prion diseases. While rare, it has been found in one to two out of 1 million cases. Several subtypes of CJD exist as studies in the medical literature, as reported the national disease monitoring organizations. In this case we saw two cardinal features of CJD including rapidly progressive mental deterioration and myoclonus. Myclonus can be provoked by startle and is present in 90 percent of cases, even when severe dementia coexists. Definitive diagnosis remains brain biopsy, performed post‐mortem. Diagnostic criteria for probable CJD, as established by the Center for Disease Control and Prevention (CDC) include:
progressive dementia
-
myoclonus or two of the following: visual/cerebellar disturbance; pyramidal/extrapyramidal dysfunction; akinetic mutism
-
Atypical EEG (periodic sharp wave complexes) and/or positive 14‐3‐3 CSF assay with a clinical duration to death less than two years, and/or MRI high signal abnormalities in caudate nucleus and/or putamen on diffusion weighted imaging (DWI) or fluid attenuated inversion recover (FLAIR) and
-
Routine investigations not suggestive of an alternative diagnosis
Conclusions:
CJD is an uncommon condition that is often misdiagnosed. Patients sometimes present to a Psychiatric unit with symptoms of conversion disorder. The gold standard for diagnosis is brain biopsy. However, with a high index of suspicion, the correct diagnosis can be made, and appropriate management initiated. As it stands, we are still searching for a cure, and it is our hope one can be found for this uncommon disease. (References are available.)