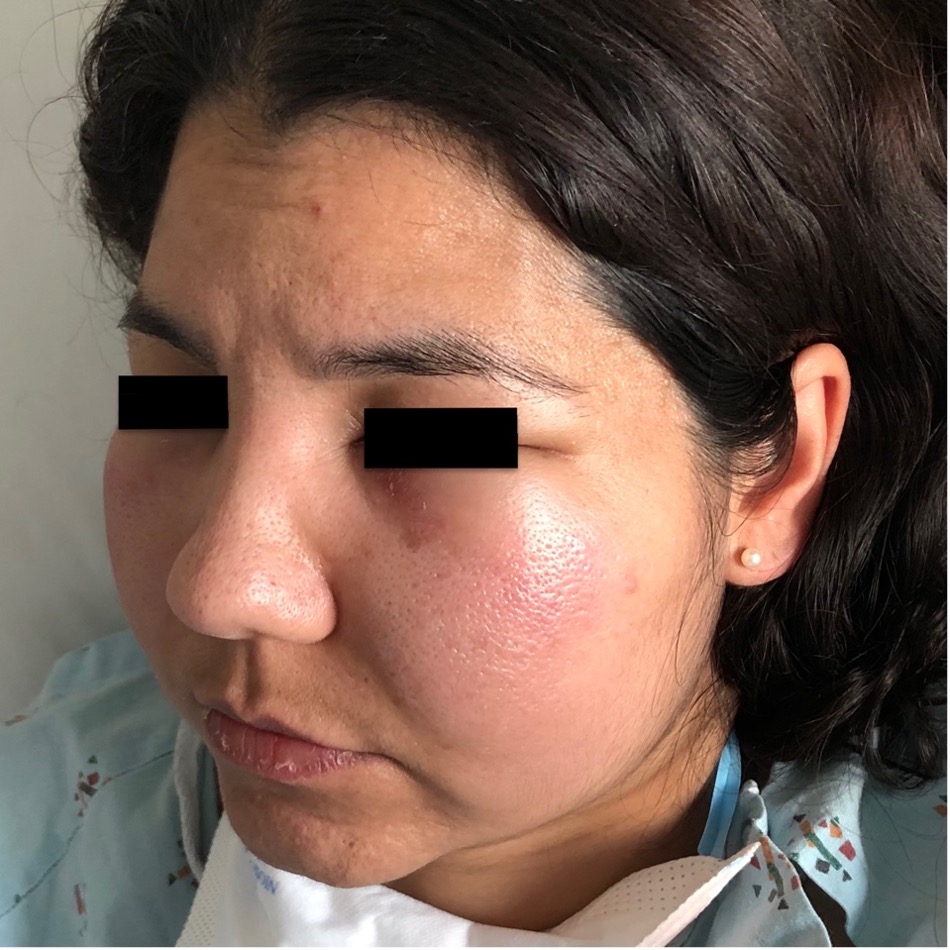
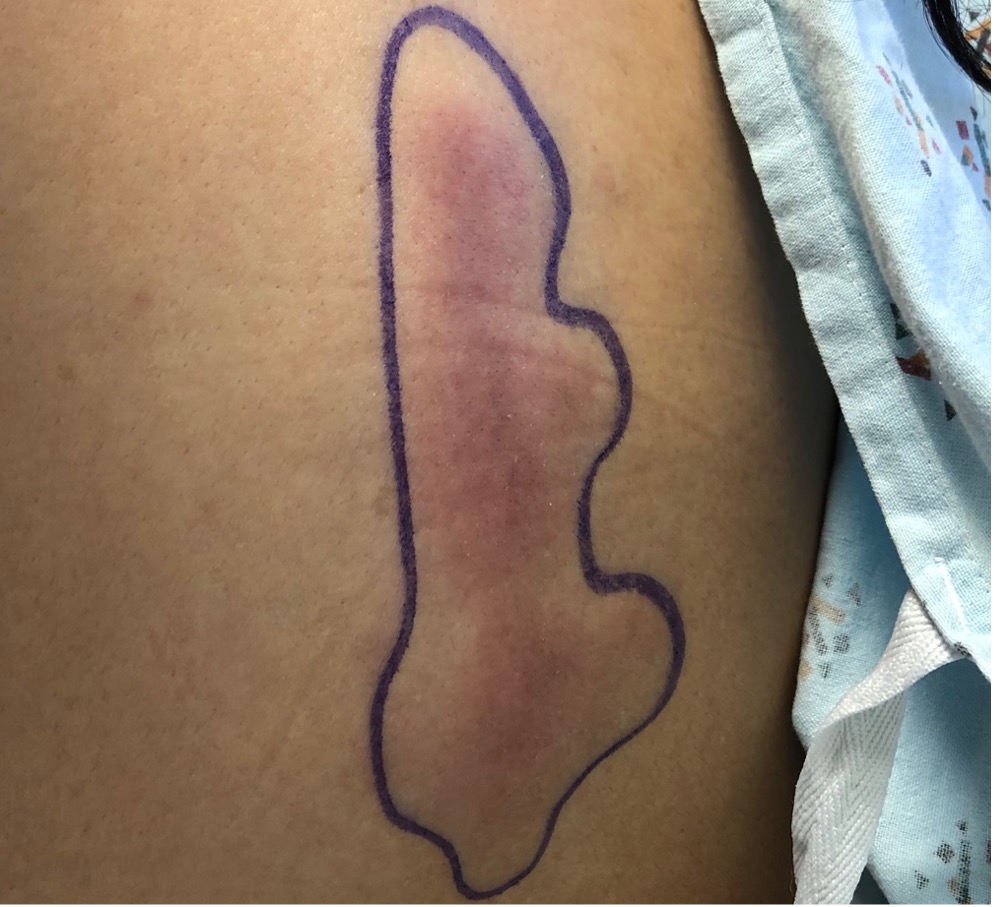
Case Presentation: A 42-year-old woman with no past medical history presented to the emergency department with four days of facial swelling and progressive, painless skin lesions on her back and extremities. Six months prior to presentation, the patient reported dark patches on her bilateral lower extremities while experiencing fever and chills. A skin biopsy was performed, and the patient was initially diagnosed with erythema nodosum. The rash persisted so the patient visited another physician who prescribed a two-month course of Rifampin and prednisone for an unclear diagnosis. The lower extremity lesions worsened and spread to her back. The patient sought medical care again and coccidioides IgM antibodies were equivocally positive. She was given oral fluconazole, but symptoms continued. Upon presentation, vital signs were significant for a temperature of 39.3˚C and a pulse of 156 BPM. Physical exam revealed bilateral indurated facial swelling and multiple painless, indurated, erythematous patches on bilateral lower extremities and torso. Labs showed hemolytic anemia, leukopenia with neutropenia, and elevated inflammatory markers (CRP 84 and LDH 590). The patient was admitted for systemic inflammatory response syndrome and fever of unknown origin. Broad spectrum antibiotics were initiated, and dermatology performed a biopsy of the rash. Repeat testing for bacterial, fungal, and viral infections was negative. Rheumatology was consulted and the patient restarted prednisone for suspected vasculitis versus cutaneous lupus. Prednisone was discontinued after negative autoimmune serologic testing. A CT scan of the chest and abdomen showed subcutaneous fat stranding and no lymphadenopathy. Hematology was consulted due to the anemia and neutropenic fever. Skin biopsy with immunohistochemistry stains revealed atypical lobular panniculitis consistent with subcutaneous panniculitis-like T-cell lymphoma (SPTCL). Treatment was initiated with cyclophosphamide, doxorubicin, vincristine, and prednisone.
Discussion: SPTCL is a rare cutaneous non-Hodgkin’s lymphoma composed of cytotoxic alpha-beta T cells that appears similar to panniculitis. SPTCL presents with an indolent course of recurrent subcutaneous painless plaques and nodules on the extremities and trunk and systemic signs and symptoms such as fever, leukopenia, and anemia. SPTCL is challenging to diagnose due to its lack of lymph node involvement and mimics other conditions such as dermatitis, erythema nodosum, or lupus. Patients’ neutropenic fever, anemia, and SIRS require hospitalization for evaluation and treatment. A multi-disciplinary approach with hospital medicine, hematology, dermatology, and pathology is essential for timely diagnosis and treatment.Diagnosis of SPTCL is based on skin biopsy and immunohistochemistry staining patterns. Dermatopathologists finalized the diagnosis. No standard treatment approach exists for SPTCL. Chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone is an effective treatment with remissions rates of up to 50%.
Conclusions: SPTCL is a rare lymphoma with an indolent progression that mimics other diseases. This patient had numerous clinical encounters and misdiagnoses before an accurate diagnosis was obtained from a skin biopsy. It is prudent that hospitalists recognize the clinical clues of SPTCL and involve dermatopathologists and hematologists for urgent skin biopsy with appropriate immunohistochemistry stains.