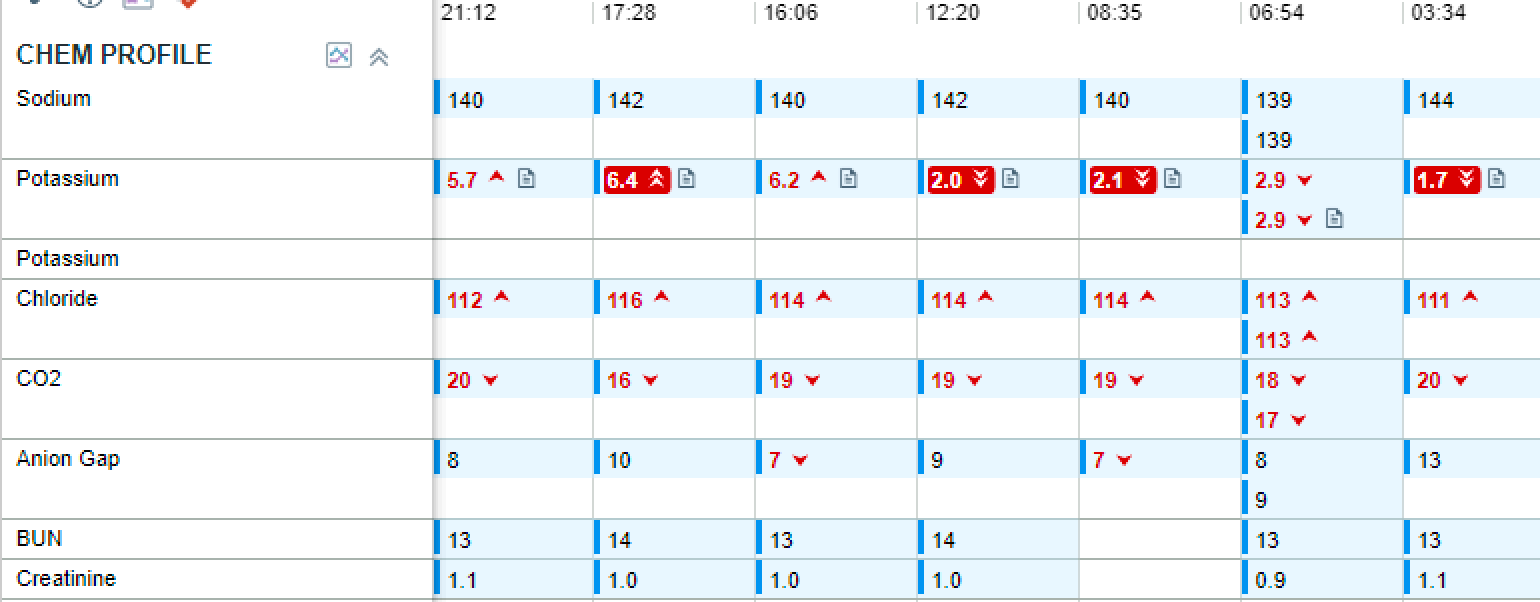
Case Presentation: An 18-year-old male presented with ascending paralysis, starting in the lower extremities and progressing to the upper extremities over 24 hours after a carbohydrate rich meal. Other review of systems was negative. He reported one prior ICU admission for similar symptoms, but details were not available. Father and grandfather had similar presentations of less severity. On physical exam, he was alert and oriented, cranial reflexes were intact. All tendon reflexes were 0/4, strength was 0/4 in the lower and 1/4 in the upper extremities, and sensation was preserved throughout. Initial labs were remarkable for potassium of 1.7 mmol/L, magnesium of 1.6 mg/dL. EKG revealed inverted T waves and prominent U waves. TSH was 2.03 µIU/mL. Urine potassium/creatinine ratio was 0.6. Patient was admitted for management of suspected hypokalemic periodic paralysis (HPP). After PO KCl 40 mEq and IV 60 mEq, hyperkalemia of 6.2 mmol/L ensued although quickly resolved. There was improvement in grip, and wiggling of toes after 6 hours when the serum K+ normalised, and he regained full function over the next 24 hours. Genetic test revealed a mutation in the SCN4A gene. Patient was discharged on eplerenone and potassium supplements.
Discussion: HPP can be familial or acquired, with a prevalence of 1 in 100,000 people. (1) Most familial cases are due to mutations in calcium or sodium channels, with autosomal dominant inheritance patterns and incomplete penetrance, especially in females. (1) Typically, acute episodes occur suddenly and are episodic, often preceded by carbohydrate intake, stress or excercise. Classic presentation includes generalized severe muscle weakness (proximal greater than distal) and a markedly low serum potassium level (less than 2.5 mmol/L). Bulbar, ocular, and respiratory muscles are usually spared, but when involved can prove fatal. Hypokalemia in these cases is secondary to redistribution, which in our case was supported by a urine potassium: creatinine ratio of less than 2.5. Rapid rebound hyperkalemia can occur even with careful repletion and requires close monitoring.As hospitalists, when evaluating ascending paralysis we should consider a broad differential and subtle clues can be key. For example, Guillain-Barre syndrome is non-episodic and has a gradual progression. Thyrotoxic periodic paralysis may mimic HPP, but occurs in hyperthyroid state. Normokalemic or hyperkalemic periodic paralysis have also been described, but are not associated with the typical HPP triggers and initial laboratories will differ. Myasthenia gravis tends to involve smaller muscles first and improves with rest. Similarly, other differentials should be considered with close attention to differences as described above.
Conclusions: Although rare, HPP should be suspected in the setting of sudden flaccid muscle weakness, especially if involving proximal muscles, prior episodes, family history or with suggestive laboratories. Initial evaluation should include thyroid function tests, and confirmation can be pursued through specialize testing. Management during acute episodes hinges on cautious potassium repletion and monitoring for hyperkalemia. Long term management focuses on educating patients about triggering factors and lifestyle modifications, although can include other measures. As hospitalists, we should remain vigilant during initial evaluation to consider uncommon conditions and, importantly, be mindful of some of their unique intricacies to avoid untoward consequences for our patients.