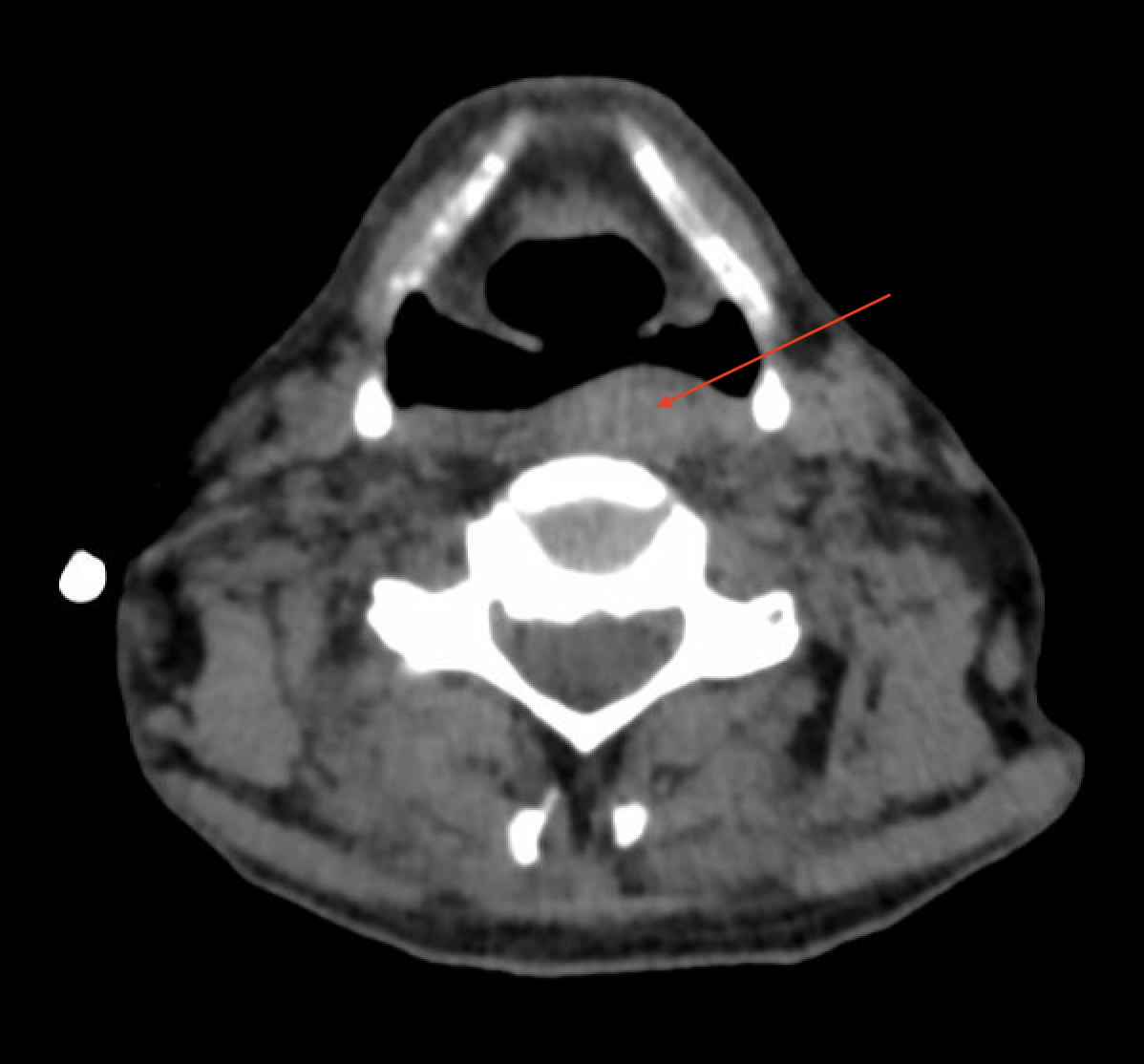
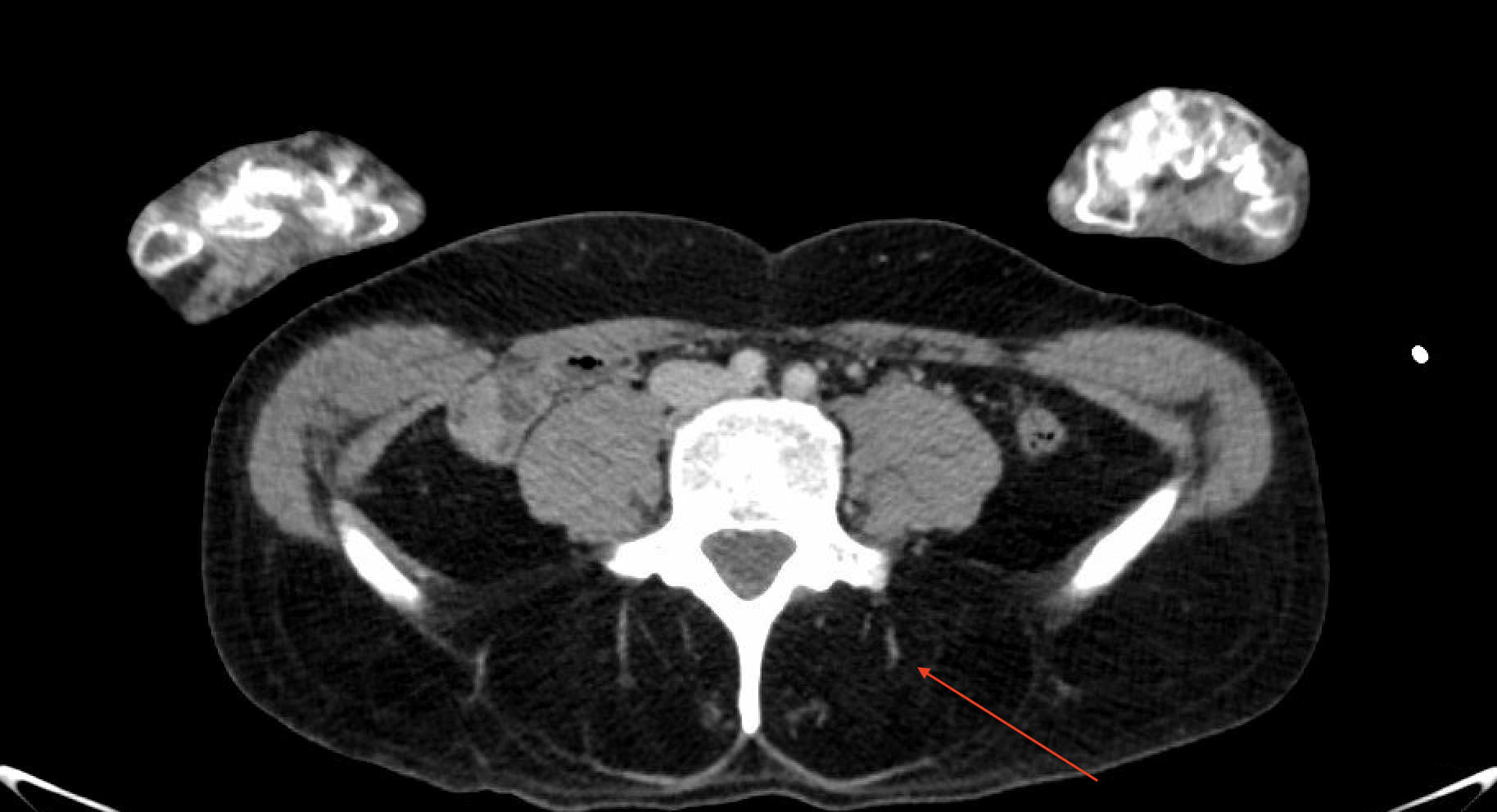
Case Presentation: A 48-year-old male with major depressive disorder, bipolar disorder and borderline personality disorder with multiple psychiatric admissions was admitted to the hospital for sepsis secondary to aspiration pneumonia with associated chronic dysphagia and unintentional weight loss. He also reported suicidal ideation, homicidal ideation, and intrusive thoughts of hurting others. Per family he had not left his apartment in over two years.His weight loss and dysphagia were suspected to be related to undiagnosed malignancy or psychiatric illness. No evidence of malignancy was found on laboratory evaluation or imaging including Computed Tomography (CT) of the neck, chest, abdomen, and pelvis. The scans revealed a parathyroid adenoma (Figure 1) and fatty replacement of paraspinal muscles(Figure 2) which was attributed to poor nutritional status. However, his neurologic exam was notable for hyperreflexia of lower extremities and hyporeflexia of the upper extremities. He was noted to have foot drop, limited function of eyelid muscles, and muscle atrophy in his bilateral upper extremities. These abnormalities created a differential diagnosis including myopathy, amyotrophic lateral sclerosis, or cervical pathology. His electromyography was consistent with type 1 myotonic dystrophy showing positive sharp waves followed by negative spikes. Final genetic testing was positive for DMPK gene with >150 repeats. Eventually, he developed hypoxic respiratory failure requiring intubation and transfer to the medical intensive care unit. He was briefly extubated, however required reintubation due to worsening hypoxia. Subsequently, he decided to defer a tracheostomy and was transitioned to comfort care.
Discussion: Muscular dystrophies are inherited disorders presenting with nonspecific symptoms and muscle weakness. Congenital myotonic dystrophy is a disorder due to CTG trinucleotide repeat expansion in the DMPK gene. Symptoms usually present earlier in life with more rapid progression as the gene is passed on. There are few reports of newly diagnosed myotonic dystrophy in middle age. For myotonic dystrophy type I symptoms include myotonia, distal muscle weakness and impairment of fine motor tasks [1]. Here we present a case of a 48-year-old male with nonspecific presentation later diagnosed with type I myotonic dystrophy. This case raises several learning points for hospitalists. His initial presentation was clouded by his psychiatric history and active infection. In hindsight his constellation of symptoms when combined with fatty replacement of paraspinal muscles seen on imaging were concerning for a neuromuscular disorder. In addition, parathyroid adenomas can be seen in myotonic dystrophy [2]. This case was challenging as patients with myotonic dystrophy usually have a family history and associated anticipation often resulting in easier and earlier diagnosis, however this was not the case for this patient. In addition, his psychiatric and social situation made obtaining an accurate history difficult. He was likely the first to have this pathogenic gene. The outcome of this case is also characteristic. Many patients eventually develop profound weakness resulting in dependence on positive pressure life support and respiratory failure usually results in death.
Conclusions: This case highlights a unique case of myotonic dystrophy presenting later in life and emphasizes the importance of maintaining broad differentials and performance of thorough physical exams in the hospital setting.