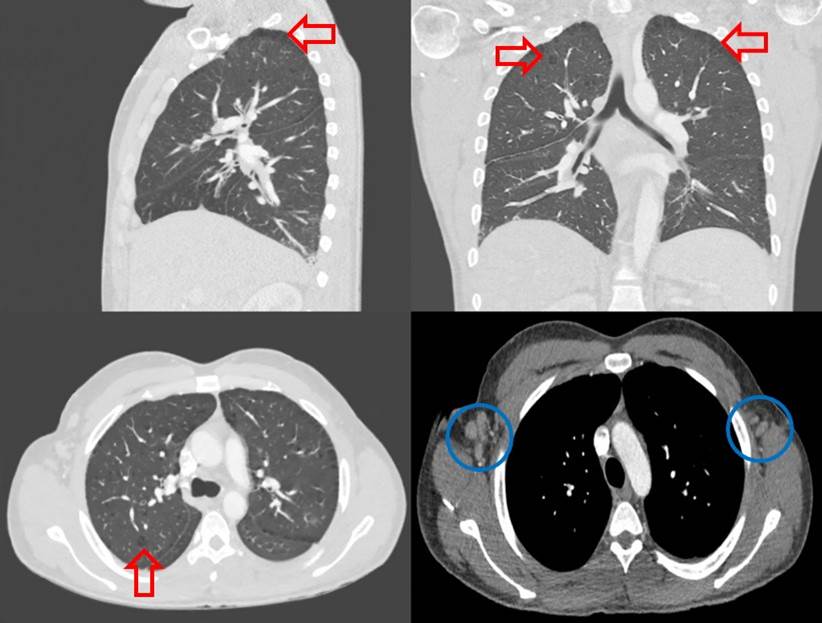
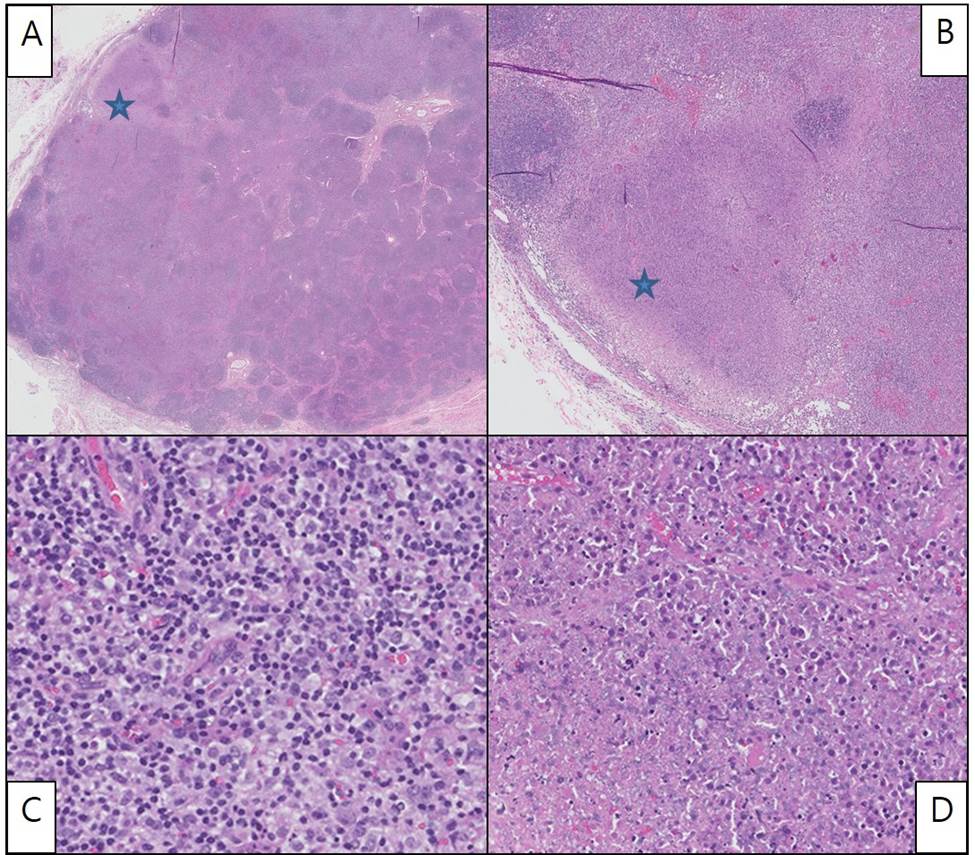
Case Presentation: A 32 year-old African American man with a history of Kikuchi-Fujimoto’s Disease (KFD), presented with 2 weeks of fever, cervical lymphadenopathy and cough. Laboratory tests showed pancytopenia, elevated ESR, positive double stranded DNA and ANA titer. Complement 3 and 4 levels were low. His fever persisted despite broad spectrum antibiotics. Computed tomography (CT) showed multiple lymph node enlargements in cervical and retroperitoneal areas and cavitary lung nodules. Tuberculosis was ruled out with three negative acid-fact bacilli (AFB) staining, culture and interferon-gamma release assay. Hematoxylin and eosin stain of axillary lymph node showed paracortical, well circumscribed necrotic lesions with abundant nuclear debris, expanded sinusoids and histiocytic proliferation with focally preserved lymph node architecture composed of monocytoid looking polyclonal (polyclonality by immunohistochemistry and by flow cytometry) B and T cells. No intermixed neutrophils or hematoxylin bodies are identified. Immunostains highlight CD68 (histiocytes), BCL6 (germinal center), PAX5 (B cells), CD20 (B cells), CD3 (T cells), CD30 (occasional immunoblots), S-100 (Rare Langerhans cells) and BCL2 (negative germinal center), whereas BCL1, EBER, GMS, AFB, BCL- 1, CAM5.2, AE1/AE3 are negative. This immuno-histologic pattern is consistent with histiocytic necrotizing lymphadenitis (KFD) especially with absence of hematoxylin bodies. SLE was also suspected because 7 out of 11 criteria were met (malar rash, oral ulcer, ANA+, arthritis, dsDNA+/antiSmith+, proteinuria, pancytopenia). Decreased levels of C3 and C4 with increased ESR were highly supportive of acute lupus flare. Methylprednisolone 30 mg IV twice a day was started for lupus flare and oral hydroxychloroquine was continued. A week after treatment, WBCs normalized and hemoglobin and platelets increased to normal range. He improved clinically with complete resolution of cough and fever. The patient was discharged with tapering regimen of high dose oral steroid with hydroxychloroquine. Outpatient follow up with rheumatology and pulmonary was recommended.
Discussion: KFD typically affects young women less than 40 years old in East Asia, but it has been reported in other races.[1] Patients with KFD present with fever and tender cervical lymphadenopathy. Other symptoms are myalgia, arthralgia, weight loss, and hepatosplenomegaly. Diagnosis can be made with excisional lymph node biopsy showing necrosis with histiocytic infiltrates.[2] 13% to 25% of KFD are associated with SLE.[3,4] Etiology is unknown, but it has been suggested that increased apoptotic pathways in KFD could accelerate the formation of autoantibodies and may subsequently cause SLE flare.[8] Characteristic morphology and histopathology of necrosis with histiocytic infiltrates in the resected lymph nodes can distinguish KFD from lupus lymphadenitis. Distinguishing SLE from KFD or even lymphoma will be critical to decide on appropriate treatment.
Conclusions: Kikuchi-Fujimoto Disease (KFD) is a self-limited disease associated with cervical lymphadenopathy and fever.[2] Herein, we report a young African American male with Kikuchi-Fujimoto disease who later presented with SLE manifestations. KFD is a rare disease. If SLE manifestations are also present, then it may be challenging to make an accurate diagnosis. Therefore, physicians need to consider KFD and SLE when a patient with fever and lymphadenopathy shows SLE manifestations.