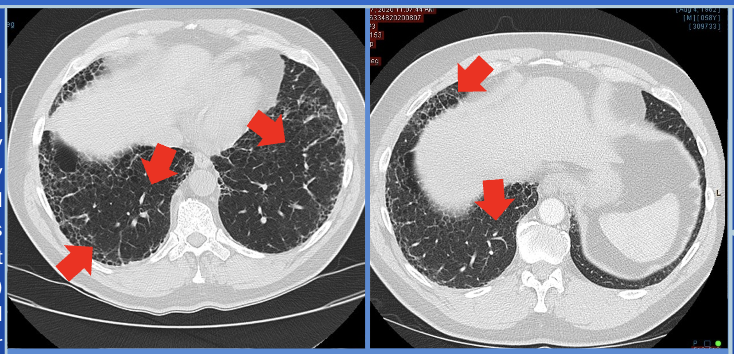
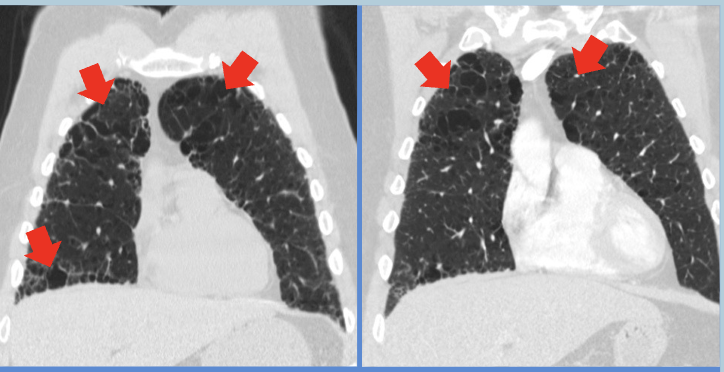
Case Presentation: Microscopic polyangiitis (MPA) is a rare form of small-vessel vasculitis strongly associated with antineutrophil cytoplasmic antibodies (ANCA), particularly myeloperoxidase (MPO-ANCA). While the kidneys are the most commonly affected organ, MPA can also present with pulmonary involvement, manifesting as interstitial lung disease (ILD). This presentation is particularly challenging to diagnose in older adults, as it mimics idiopathic pulmonary fibrosis (IPF) in its clinical and radiological patterns. Unlike other pulmonary manifestations of MPA, such as alveolar hemorrhage or non-specific interstitial pneumonia (NSIP), cases of MPA-related ILD frequently follow a gradual course of symptom progression. This report describes an elderly male presenting with progressive ILD and positive P-ANCA/MPO, which led to the diagnosis of MPA.A 62-year-old male with a 48-pack-year smoking history and occupational exposure to fumes and dust presented with progressive dyspnea and chronic cough, which worsened over the past year. These symptoms were noted despite ongoing treatment for a prior diagnosis of chronic obstructive pulmonary disease (COPD). Incidentally, a low-dose computed tomography (LDCT) of the chest revealed interstitial lung disease (ILD).On physical examination, the patient appeared in no acute respiratory distress. Auscultation findings were normal, and vital signs, including heart rate, respiratory rate, and oxygen saturation, were stable. A thorough systemic evaluation, including cardiovascular, respiratory, abdominal, cranial, and skin examinations, showed no abnormalities. A CT scan of the chest demonstrated significant progression of fibrosis predominantly in the lower lobes, with findings such as honeycombing and traction bronchiectasis suggestive of a usual interstitial pneumonia (UIP) pattern.While the patient reported a history of gastroesophageal reflux disease (GERD), it was well-controlled with proton pump inhibitors (PPI) and unlikely to contribute to the observed lung pathology. Secondary workup for UIP was initiated, including autoimmune markers and hypersensitivity panels. This workup revealed positive P-ANCA and myeloperoxidase (MPO) antibodies, alongside elevated inflammatory markers like erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). These findings confirmed the diagnosis of MPA-associated ILD.The diagnosis underscores the importance of differentiating MPA-ILD from IPF, as the treatment approaches differ significantly. Early diagnosis is critical, as immunosuppressive therapy can effectively manage MPA and halt disease progression, unlike the largely supportive care required for IPF.
Discussion: ILD is increasingly detected on LDCT, as in this case. Identifying the cause of progressive fibrotic lung diseases is crucial to prevent worsening ILD. MPA with pulmonary involvement is often acute or subacute, such as DAH or NSIP, but is less recognized for fibrotic presentations like UIP. MPO-ANCA testing is key for early diagnosis and distinguishing MPA from IPF. Early detection allows immunosuppressive treatment to slow ILD progression. Routine ANCA testing is essential in unexplained ILD, as MPA and IPF share similar radiological findings.
Conclusions: This case highlights the importance of early diagnosis of MPA in patients with ILD. Routine MPO-ANCA testing is essential to differentiate MPA from IPF, enabling appropriate treatment to slow disease progression.