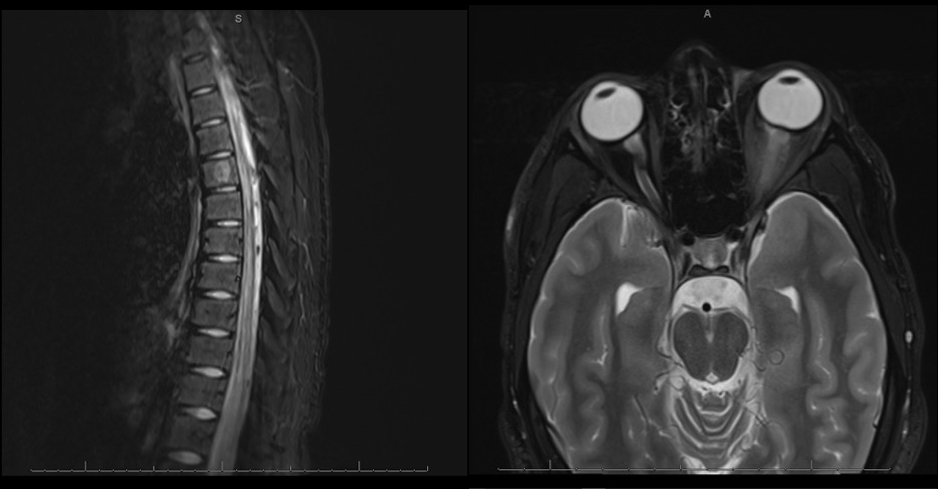
Case Presentation: A 17-year-old healthy male teenager presented with 2 weeks of bilateral lower extremity weakness with paresthesia, urinary retention, blindness of the left eye, left relative afferent pupillary defect, and retained deep tendon reflexes. An MRI of the orbit, brain, and spine showed left optic neuritis and transverse myelitis extending from T9 to the conus medullaris. Cerebrospinal fluid (CSF) analysis showed nucleated cells at 250, protein at 48, lymphocytes at 98, and glucose at 62. Comprehensive infectious workup for HSV, enterovirus, Lyme, West Nile, syphilis, HIV, HTLV-1, and EBV was negative. Serum and CSF antibody testing showed no anti-AQP4 antibodies, but it was positive for the anti-MOG antibody in serum. He was treated with a consecutive course of high-dose IV methylprednisolone and plasmapheresis. He experienced return of strength in his lower extremities, improvement in assisted walking, and improvement of vision with treatment. He was discharged on a prolonged steroid taper and close follow up with neurology.
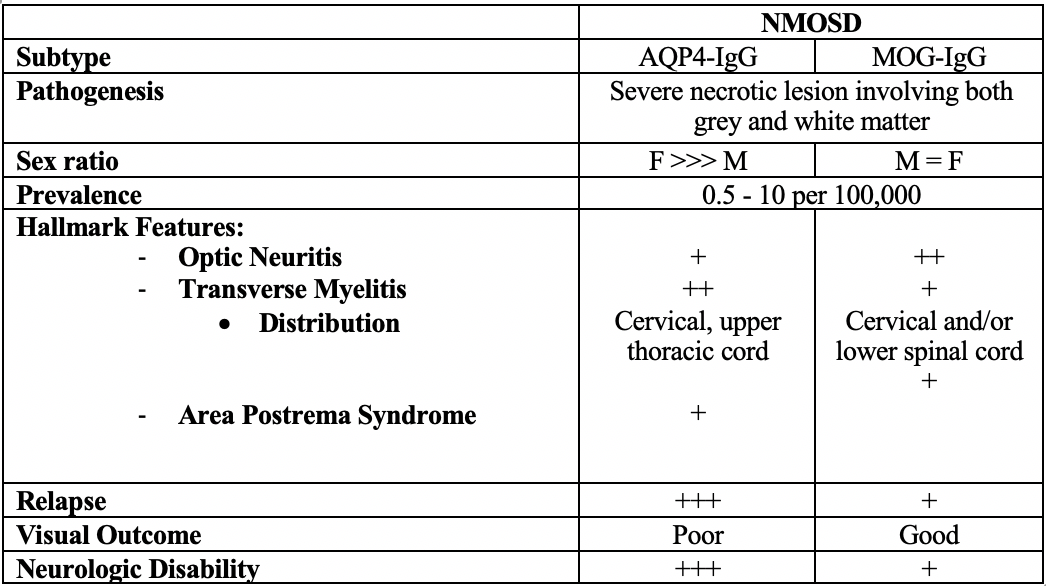
Discussion: Neuromyelitis Optica Spectrum Disorder (NMOSD) is an autoimmune demyelinating disease of the central nervous system (CNS) presenting with optic neuritis and transverse myelitis (1). The presumed pathogenesis of NMOSD is mostly due to autoantibodies against the aquaporin-4 channel (anti-AQP4 antibodies). Recent research has found antibodies against myelin oligodendrocyte glycoprotein (anti-MOG) which has a distinct clinical pattern in patients who are anti-AQP4 seronegative (2,3). We present an interesting pediatric case of MOG antibody disease.Anti-MOG driven NMOSD has an emerging distinct clinical picture notable for greater optic nerve involvement, lower relapse rates, longitudinal lesions, and a greater occurrence in males when compared to classic NMOSD (2,3). The literature lacks evidence on the most effective treatments and relapse prevention for the disorder, particularly in pediatric patients due to the rarity of the disease (3).Patients with optic neuritis due to anti-MOG antibodies may have a more favorable outcome for vision than those who are anti-AQP4 seropositive (3,7). Limited studies suggest anti-MOG NMOSD may have a greater response to steroids than AQP4 NMOSD, thus prolonged steroid tapers are preferred (3,6) Further research into long-term therapy for relapse prevention with immunosuppression is needed to determine the best treatment course for anti-MOG NMOSD (8).
Conclusions: Clinicians should be suspicious of an autoimmune etiology after ruling out infectious causes in patients presenting with optic neuritis, progressive weakness, urinary retention, and imaging findings of extensive inflammation (3,4). The serum anti-MOG antibody test has a reported specificity of 98.5% for anti-MOG associated disease (5).The exact distinction between anti-AQP4 and anti-MOG NMOSD is still being explored in the current literature. However, it is clear that anti-MOG-mediated disease has a diverging presentation consistent with lower rates of relapse, greater optic nerve involvement, a loss of female predominance, and better overall outcomes when compared to anti-AQP4 NMOSD.