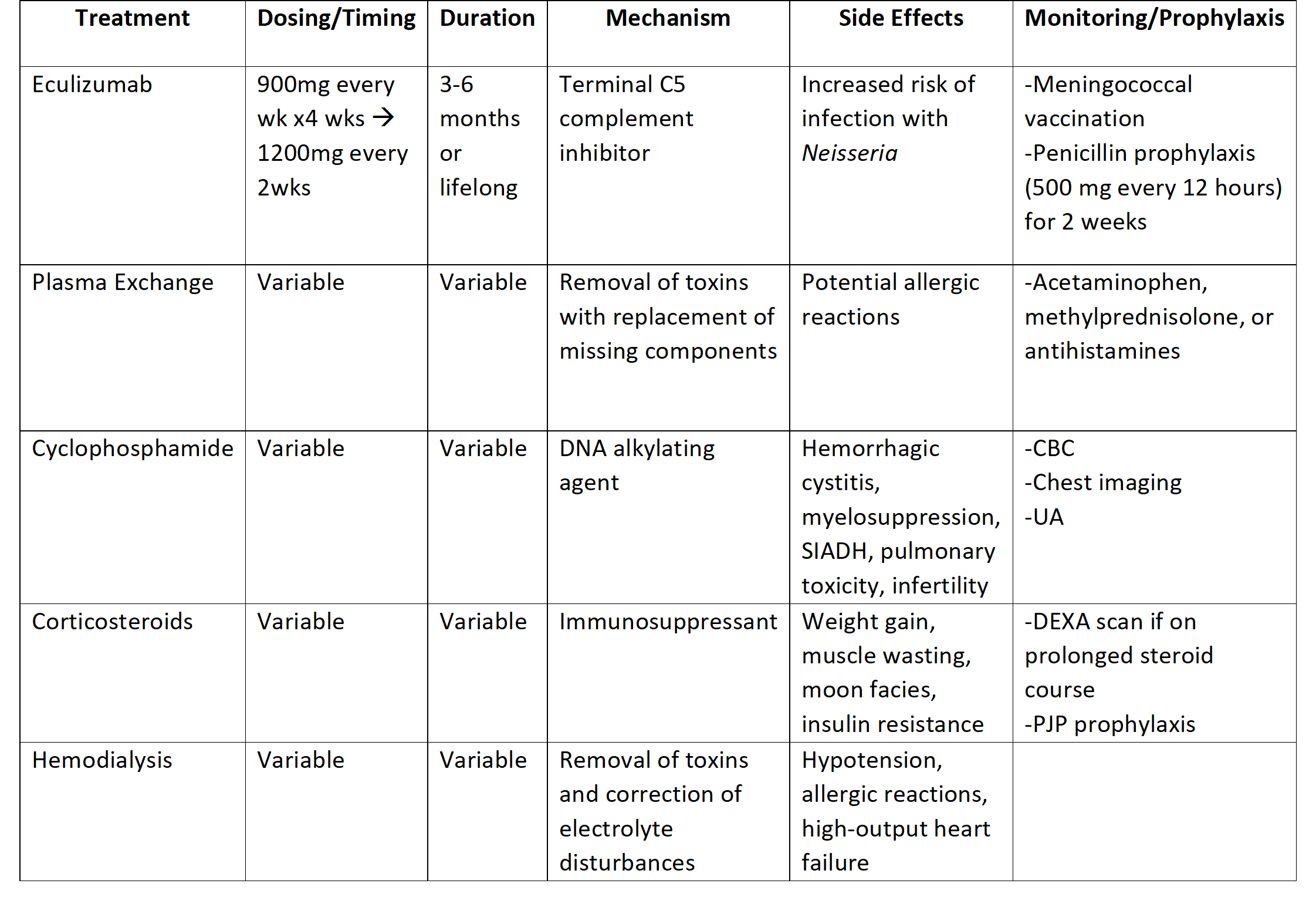
Case Presentation: The patient is a 27-year-old male with lupus anticoagulant with hypoprothrombinemia syndrome (LAHPS)/triple positive antiphospholipid syndrome (APLS) in the setting of systemic lupus erythematous (SLE) and immune thrombocytopenia (ITP) on hydroxychloroquine, mycophenolate mofetil (MMF), and prednisone for immunosuppression, who presented to the ER with worsening right upper quadrant and epigastric pain. RUQ US and HIDA scan were concerning for chronic cholecystitis. He underwent laparoscopic cholecystectomy complicated by a bile leak. Post operatively, CT A/P showed small hepatic and gallbladder fossa fluid collections, so MMF held due to suspected GI infection. He later developed diarrhea, but C. difficile infection and bile acid diarrhea were excluded, narrowing the differential to MMF as the most likely cause of diarrhea. He then developed worsening kidney function. CBC revealed anemia and thrombocytopenia, and schistocytes were noted on smear. Renal biopsy showed mild mesangial immunocomplex deposition consistent with mild class II lupus nephritis. Interstitial fibrosis and tubular atrophy (IFTA) score were 10%, which suggested limited chronic disease and raised suspicion for autoimmune TMA associated with SLE and APLS (specifically atypical hemolytic uremic syndrome, or aHUS). Negative ADAMTS13 and factor H autoantibody ruled out TTP and aHUS, respectively. Studies also revealed low complement levels and elevated beta 2 microglobulin, anticardiolipin and anti-dsDNA levels, making autoimmune TMA secondary to SLE and lupus nephritis flare the most likely diagnoses. Due to his worsening non-oliguric kidney failure, patient began short term hemodialysis and weekly eculizumab infusions for treatment of autoimmune related TMA.
Discussion: Thrombotic microangiopathy (TMA) represents a broad group of systemic disorders that are characterized by thrombocytopenia, microangiopathic hemolytic anemia, and renal dysfunction. Primary causes of TMA include thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome.1 Secondary TMAs commonly arise in conjunction with HELLP syndrome, organ transplantation, malignancy, autoimmune conditions, and medications. Autoimmune diseases associated with TMA include SLE, systemic sclerosis, antiphospholipid syndrome, Hashimoto thyroiditis, and vasculitides.2 The most common autoimmune disorder thought to provoke TMA is SLE. Antiphospholipid syndrome (APLS) is another autoimmune condition associated with secondary TMA. 1 Treatment of autoimmune TMA secondary to SLE or APLS has centered around treating the underlying SLE or APLS with corticosteroids, hemodialysis, and anticoagulation. However, eculizumab, plasma exchange therapy, mTOR inhibition, and B cell depleting therapy have been shown beneficial in treatment of autoimmune TMA.3
Conclusions: Autoimmune TMA is a diagnosis of exclusion, as its presenting symptoms are similar to other types of TMA, such as thrombotic thrombocytopenic purpura and typical/atypical HUS. Recognition and diagnosis through multi-disciplinary approach in hospital medicine are heavily associated with better patient outcomes, but the diagnosis can be difficult due to its nonspecific presentation. This case highlights the need for coordinated care from subspecialties when practicing hospital medicine, such as hematology, rheumatology, and nephrology, to reduce the morbidity and mortality of autoimmune TMA associated with SLE and APLS.