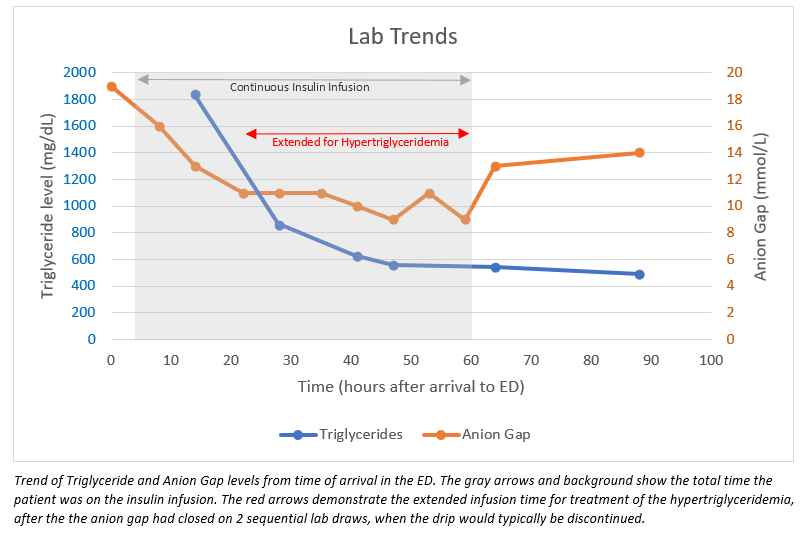
Case Presentation: A 44-year-old man with no past medical history presented to the ED with 3 days of severe epigastric abdominal pain with associated nausea and vomiting. He also endorsed polyuria for 3 days and denied significant alcohol use. His lipase was elevated at 469 U/L, establishing the diagnosis of pancreatitis, which was confirmed by CT scan findings of reactive inflammation and fat stranding. Additionally, he had mild diabetic ketoacidosis (DKA), with a venous pH of 7.25, bicarbonate 15 mmol/L, anion gap 19 mmol/L, blood glucose 282 mg/dL, elevated beta-hydroxybutyrate, and ketonuria. The patient was admitted for treatment of acute pancreatitis and DKA. He was kept NPO with maintenance fluids and continuous insulin infusion. On hospital day 1, further studies to determine the etiology of his pancreatitis revealed severe hypertriglyceridemia of 1,839 mg/dL. Abdominal ultrasound was negative for gallstones. Endocrinology was consulted and recommended continuing the insulin infusion until his triglycerides (TGs) were <500 mg/dL. Therefore, he was transitioned to a subcutaneous insulin regimen approximately 60 hours after presentation, which was 38 hours after his anion gap closed on two sequential lab draws. His glycated hemoglobin resulted at 10% and TGs down trended to 489 mg/dL. He was discharged on hospital day 4 upon symptomatic improvement with fenofibrate, metformin, and Basaglar insulin.
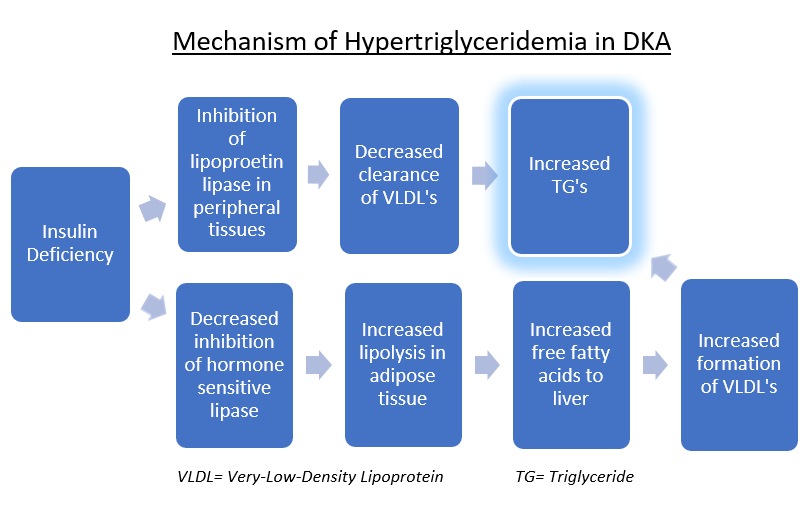
Discussion: The triad of DKA, hypertriglyceridemia, and acute pancreatitis is extremely rare in a previously undiagnosed diabetic patient (1). The mechanism is thought to originate from the insulin deficiency. Ultimately, circulating VLDL particles increase, which carry TGs. Pancreatitis occurs because the release of free fatty acids is toxic to acinar cells and capillaries, with significant risk once TG concentrations are >1000 mg/dL (1,2). Of note, a contrasting mechanism has been demonstrated by another case report, in which DKA occurred 4 days after the onset of acute pancreatitis, in the absence of hypertriglyceridemia (3). This suggests pancreatitis was the inciting factor, resulting in acute beta-cell dysfunction and resultant insulin deficiency. However, in our case the DKA was the primary event, as his glycated hemoglobin evidenced chronic hyperglycemia and a new diabetes diagnosis. Severity scoring systems such as APACHE II and Ranson criteria are unreliable predictors in these cases since many of the lab markers used are influenced by the underlying DKA. Instead, CT scan findings have been suggested to better correlate with outcomes (4). Management mainly consists of NPO status, IV fluids, continuous insulin infusion, and lipid-lowering agents. The insulin works by reversing the metabolism pathway outlined in figure 2. However, a comparison study of insulin infusion versus fasting alone suggested that insulin does not induce a greater fall in TGs (5). Additionally, trials comparing the efficacy of other treatment modalities, such as apheresis and heparin, are lacking.
Conclusions: This rare triad is important for hospitalists to recognize because the current literature supports treating hypertriglyceridemia with insulin infusion until levels fall below 500 mg/dL. If the hypertriglyceridemia is not identified, the insulin infusion could be discontinued prematurely upon closure of the anion gap, leading to continued pancreatic inflammation and the potential for additional complications and prolonged hospitalization.