Case Presentation: A 60-year-old female with a history of Castleman disease presented outpatient with painful progressive “stocking-and-glove” polyneuropathy. She was referred for EMG, which demonstrated significant sensorimotor neuropathy. Notable lab findings months prior included new subclinical hypothyroidism, normocytic anemia, and thrombocytosis. Given her presentation, she was admitted for expedited evaluation. Upon arrival, neurology and hematology were consulted. Examination was notable for whitening of the nails, left supraclavicular lymphadenopathy, upper and lower extremity numbness, decreased plantarflexion, and decreased grip strength. Significant lab findings included elevated VEGF and IgG lambda monoclonal protein. A PET scan showed increased size and metabolic activity in the left supraclavicular lymph nodes, worsening of known lytic lesions, and new lytic lesions in the ribs and C7-L1 vertebrae. MRI of the spine revealed a T5-T7 lobulated mass and marrow-replacing lesions within C7-L1 vertebrae (Figure 1A). CT-guided biopsy of the lobulated mass demonstrated a plasmacytoma (Figure 1B). Surgical cervical lymphadenectomy revealed a lambda-restricted plasma cell neoplasm. Bone marrow biopsy demonstrated a mildly hypercellular bone marrow and polytypic plasmacytosis with a subset of plasma cells with aberrant TP53 signaling.The patient was discharged with hematology and radiation oncology follow-up. Ultimately, hematology diagnosed her with Polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes (POEMS) syndrome and concurrent multiple myeloma (MM). She was treated with chemotherapy and radiation.
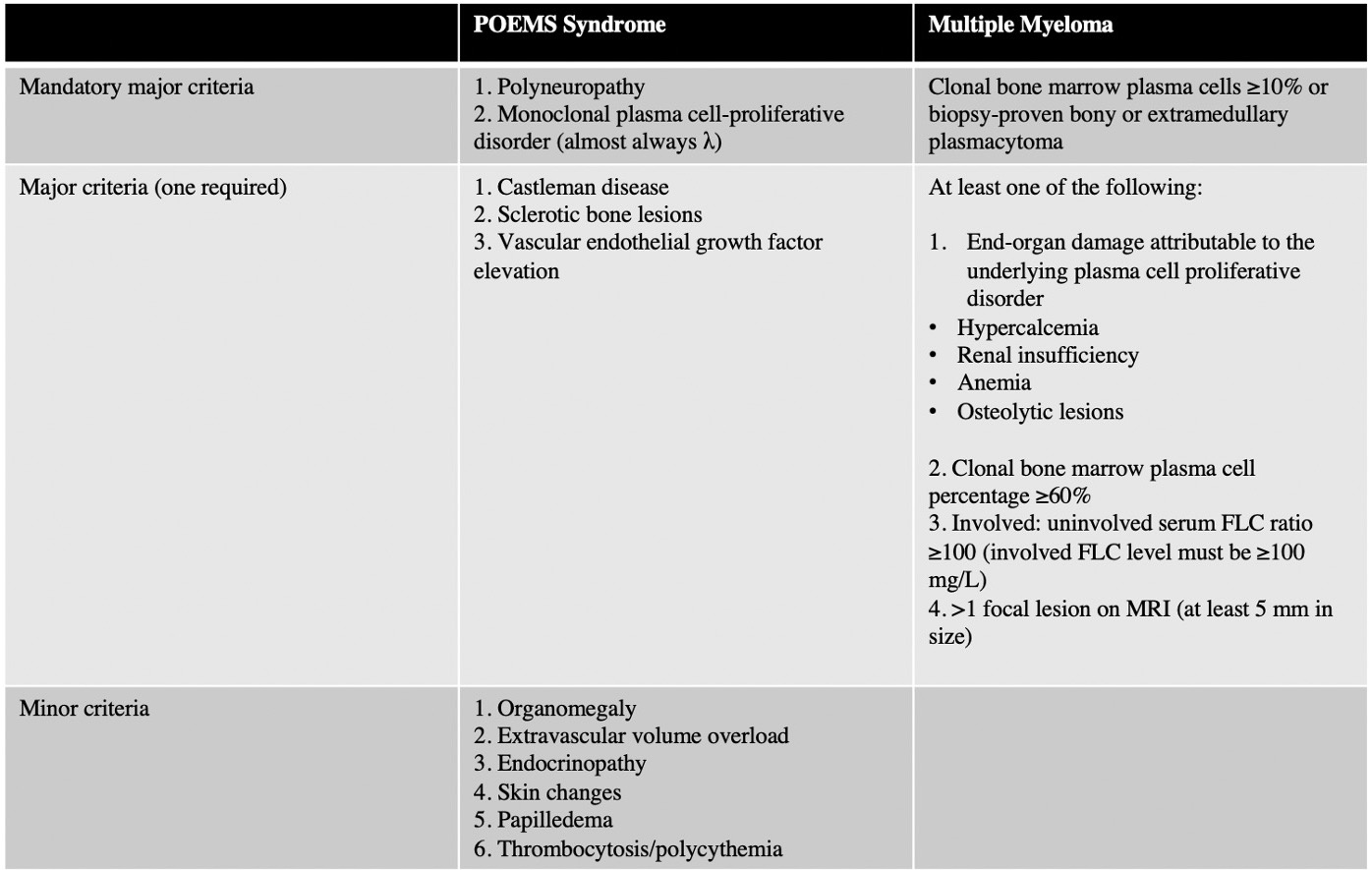
Discussion: POEMS syndrome is a rare paraneoplastic syndrome associated with Castleman disease, a heterogeneous lymphoproliferative disorder. Diagnosis of POEMS syndrome requires two mandatory criterion, polyneuropathy and a monoclonal plasma cell disorder, along with at least one other major and one minor criterion.(1) Diseases such as chronic inflammatory demyelinating polyneuropathy (CIDP), MM, monoclonal gammopathy of undetermined significance (MGUS) neuropathy, and primary systemic amyloidosis can mimic POEMS, contributing to a median time from symptom onset to diagnosis of approximately one year.(2) Because clinical manifestations of POEMS syndrome are variable, misdiagnosis or delayed diagnosis can result in inappropriate treatment and a poorer prognosis. POEMS syndrome can be differentiated from MM based on several features, including neuropathy as the dominant symptom, Castleman’s disease, VEGF elevation, and sclerotic bone lesions.(4) In this case, the patient met criteria for POEMS, meeting both mandatory criteria, all three other major criteria with sclerotic bone lesions, elevated VEGF, and Castleman disease, and multiple minor criteria with hypothyroidism, skin changes, and thrombocytosis. While the lambda-predominant monoclonal proliferative disorder overlaps with MM, the bony plasmacytoma, the aberrant TP53 signaling plasmacytosis on bone marrow biopsy, lytic bone lesions, and anemia are distinct to MM (Table 1).(5)
Conclusions: While the intersection of the two diseases is rare, it is essential to differentiate POEMS syndrome from MM for treatment and prognostic purposes. Confirming the correct diagnoses made appropriate therapy options available for this patient. To our knowledge, this is one of very few reported cases of POEMS syndrome with concomitant MM.