Case Presentation:
A 49 year old male with no past medical history of hypertension, presented to the ER for evaluation of headache associated with nausea and vomiting.
Patient reported that he developed throbbing headache over the past few days. Associated symptoms included nausea and vomiting. On further questioning, he reported that he had a spell of headache a few weeks ago, associated with palpitations and diaphoresis. He presented to the ER where his blood pressure was found to be 260/148. He had no other medical or surgical problems, nor does he smoke or use any illicit substances. Physical exam was unremarkable except for diaphoresis and tachycardia. Pertinent lab findings included a creatinine of 1.8 and mild troponin elevation at 0.4. His urine drug screen was negative. His EKG showed sinus arrhythmia with periods of junctional tachycardia. He was admitted to the hospital for Hypertensive emergency and was started on Nicardipine drip and PRN labetalol. However, his blood pressure and heart rate were labile with intermittent tachycardia up to 140`s and systolic blood pressure that fluctuated between 220`s and dropped to as low as 90`s.
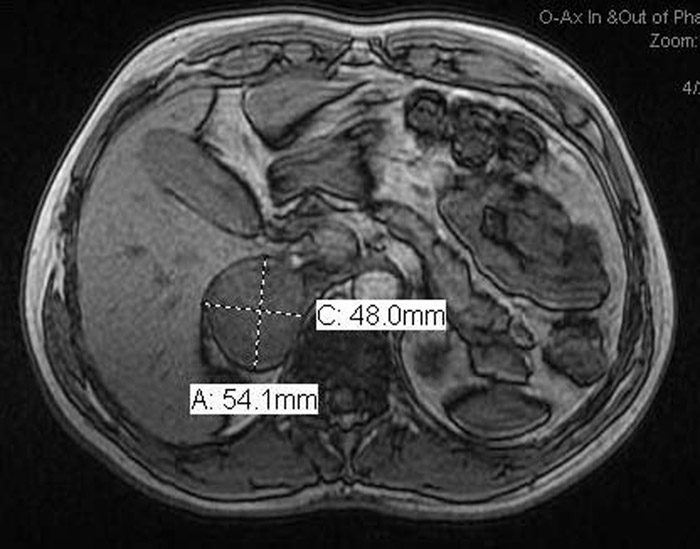
Given no prior history of hypertension, an evaluation for secondary hypertension was undertaken. His thyroid studies, Aldosterone levels were normal. A 24 hour urine and plasma metanephrines revealed markedly elevated levels, biochemically supporting a diagnosis of pheochromocytoma. A renal ultrasound revealed a 5X4 cm adrenal mass, which was confirmed with an Adrenal MRI. He was started on Doxazosin followed by Atenolol and subsequently underwent an elective right adrenalectomy. Pathology was immunoreactive for chromogranin, synaptophysin, and S‐100 immunostains confirming the diagnosis of pheochromocytoma. Three months after surgery, he is doing well and is off all blood pressure medications.
Discussion:
Pheochromocytomas are rare catecholamine‐secreting tumor that arise from chromaffin tissue within the adrenal medulla and extra‐adrenal sites. Their clinical hallmarks are sustained or intermittent hypertension often associated with paroxysmal symptoms. The classic triad of symptoms in patients with a pheochromocytoma consists of episodic headache, sweating, and tachycardia. The diagnosis is typically confirmed by measurements of urinary and plasma fractionated metanephrines and catecholamines. Biochemical confirmation of the diagnosis should be followed by radiological evaluation to locate the tumor using CT/MRI or MIBG scintigraphy. Medical management consists of alpha blockers followed by beta blockers. Beta Blockers are contraindicated as an initial therapy, as unopposed alpha activity can lead to severe hypertensive crises. Surgical resection is recommended after blood pressure is controlled. Serial biochemical monitoring is required as a small percentage of these tumors have malignant potential.
Conclusions:
Pheochromocytoma should be considered in the differential of patients presenting with hypertensive emergency and/or labile, paroxysmal hypertension. Hospitalists manage a large percentage of hypertensive emergencies and workup for secondary causes of hypertension should be considered in young patients and those with resistant hypertension. Although the diagnosis of Pheochromocytoma is often elusive; early diagnosis is important, as this changes management and prevents long term morbidity and mortality