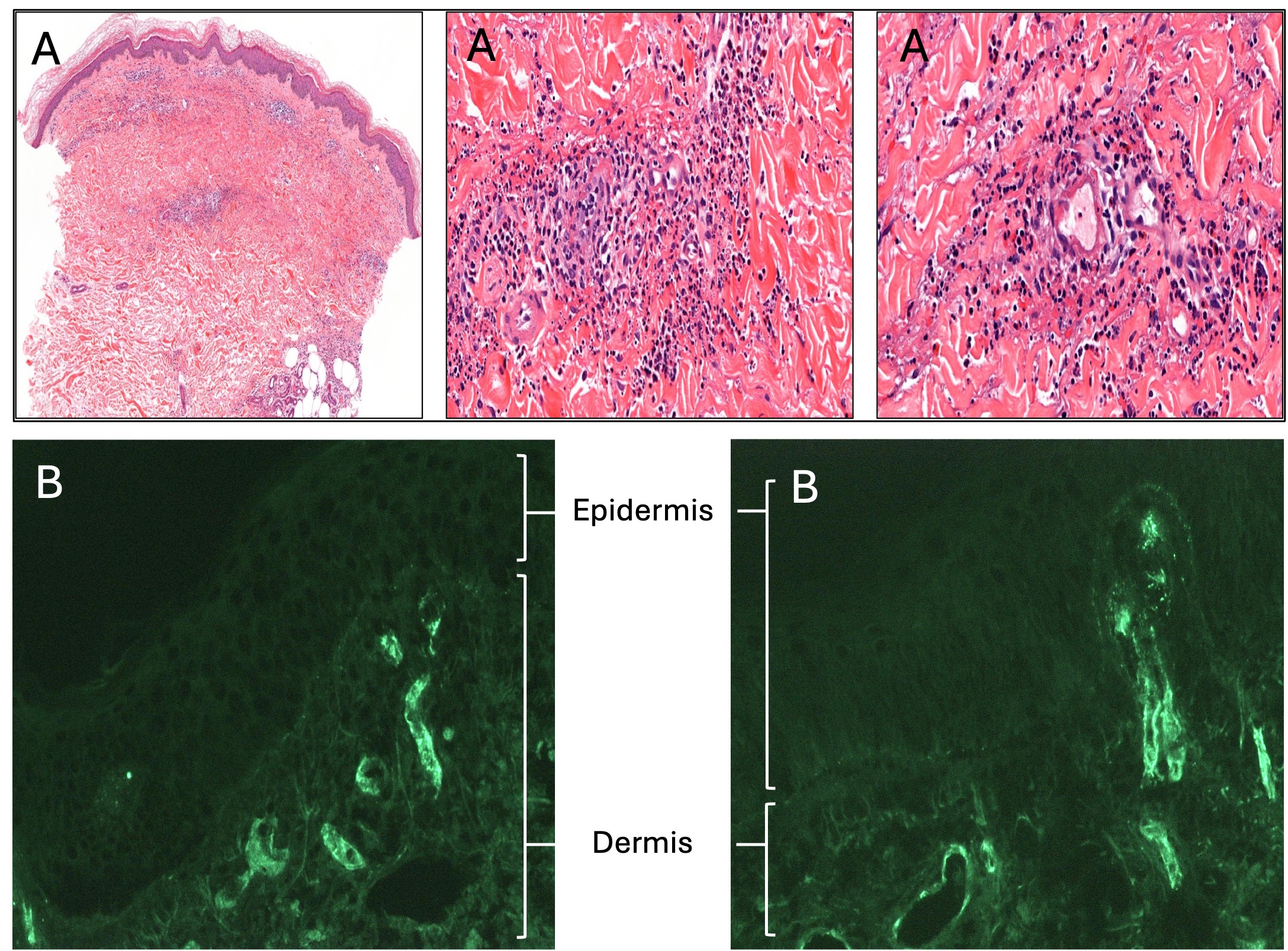
Case Presentation: A 55-year-old female with hypertension presented to the Emergency Department with bilateral lower extremity rash. Five days prior, she developed fatigue and small red spots on her distal legs which progressed to pruritic, raised, palpable lesions. Over-the-counter steroid cream reduced the pruritus, but the rash spread proximally, prompting presentation. She was hypertensive with palpable purpura from bilateral thighs to feet (Figure 1A). Labs showed increased hemoglobin (16 g/dL; reference range [RR] 11.3-14.9 g/dL), platelets (1,043×109/L; RR 150-450×109/L). Additional diagnostics included low immunoglobulin (Ig) M (25 mg/dL; RR 40 – 230 mg/dL); elevated IgA (510 mg/dL; RR 70–400 mg/dL); and normal urinalysis, ESR, serum immunofixation, rheumatoid factor, C3, C4, IgG, anti-streptolysin O, ANA, cryoglobulin, and ANCA. Peripheral smear showed thrombocytosis. Skin biopsy revealed leukocytoclastic IgA vasculitis (Figure 2). Myeloproliferative neoplasm panel identified JAK2 V617F mutation. She was diagnosed with concomitant IgA vasculitis and polycythemia vera (PV) and started ruxolitinib with improved cytoses and purpura (Figure 1B).
Discussion: IgA vasculitis presents with palpable purpura, gastrointestinal symptoms, arthralgias, and renal involvement. Often preceded by an upper respiratory infection, it frequently affects children. Diagnosis requires palpable purpura or petechiae (without thrombocytopenia) and abdominal pain, renal involvement, arthralgia or arthritis, or proliferative glomerulonephritis or IgA-predominant leukocytoclastic vasculitis. Barring renal involvement, treatment is supportive.1PV is a chronic myeloproliferative neoplasm. Patients with PV can be asymptomatic or present with headache, fatigue, dizziness, gastritis, vision changes, insomnia, pruritis, claudication, erythromelalgia, aquagenic pruritis (40%). Bleeding and thrombosis are infrequent (1%). Diagnosis requires elevated hemoglobin/hematocrit with positive JAK2 gene mutation (90%) or bone marrow with panmyelosis. Treatment focuses on reducing thrombosis, bleeding, or transformation risk. A combination of phlebotomy (target hematocrit < 45%), and low-dose aspirin reduces thrombotic risks. Hydroxyurea reduces platelet counts. A JAK inhibitor, ruxolitinib, can help achieve complete hematologic remission.2Co-presenting IgA vasculitis and PV is rare. All 13 cases in English publications describe males with IgA vasculitis renal involvement; 6 with PV before IgA vasculitis, 3 with IgA nephropathy before PV diagnosis, and 4 with concomitant diagnoses.3–8 Common findings include hypertension, hepatomegaly or splenomegaly, hematuria or proteinuria, flushing, conjunctival suffusion, visual changes, and palpable purpura.3-8 Three published cases reported positive JAK2 mutation;6,7 others were published prior to JAK2 testing.9 Our case is exceptionally rare in that the patient is female, presented with concomitant IgA vasculitis and PV, and had no renal dysfunction.
Conclusions: Given variable presentations, clinicians should be suspicious of concomitant IgA vasculitis and PV in patients with palpable purpura, renal dysfunction, hypertension, or hepatosplenomegaly. Our patient presented without renal dysfunction, suggesting early diagnosis and intervention may reduce progression to nephropathy.