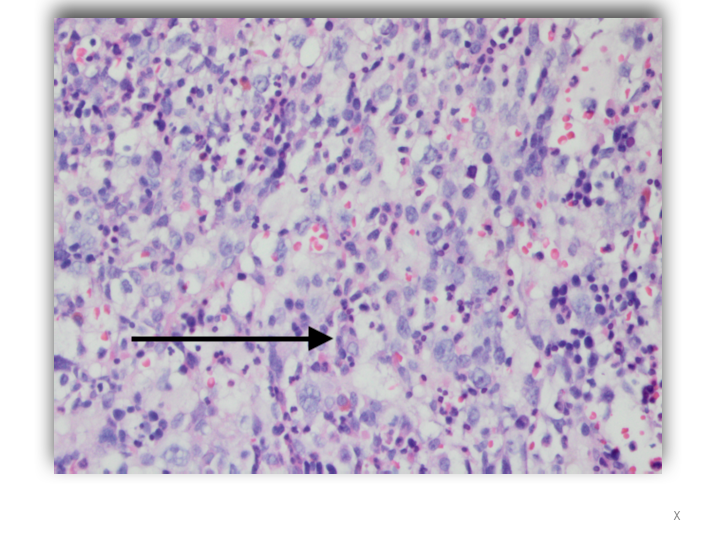
Case Presentation: A 40-year-old man presented to the Emergency Department with fatigue, progressive shortness of breath, fever and night sweats for three months. During this period, he had episodes of blurred vision and photosensitivity. A diagnosis of Vogt Koyanagi Harada uveitis was made following an ophthalmologic assessment. He was treated with prednisone 40mg daily. Physical examination revealed oral candidiasis and splenomegaly. He had no lymphadenopathy. Laboratory studies showed Hb 7.4g/dL WBC: 5.8x 103 μL, platelets 72 x103/μL, LDH 1042 u/L, ferritin 1829 ng/L, Triglyceride 619 mg/dL and lactic acid 4.3 mmol/L. Viral serology for EBV, CMV, HIV and viral hepatitis were negative. However, he was positive for Coxsackie A IgG. Thoraco-abdominal CT confirmed splenomegaly. Vancomycin and cefepime were empirically started. The patient’s hospital course was marked by continued fevers as well as severe thrombocytopenia and anemia with a nadir of 15×103/µL and 6.8 g/dl respectively, both requiring transfusions. Blood cultures returned without growth at the end of incubation. Bone marrow biopsy was done, revealing hemophagocytosis and Diffuse Large B Cell Non-Hodgkin Lymphoma. Soluble CD25 receptor level was 2156 pg/ml. Lumbar puncture showed no central nervous system involvement. Systemic chemotherapy with dexamethasone and etoposide was commenced. Thereafter his fevers and cytopenia improved. He is currently receiving intrathecal Methotrexate for CNS prophylaxis as an outpatient.
Discussion: Hemophagocytic Lymphohistiocytosis (HLH) is a rare disease characterized by hyperproliferative macrophages which phagocytose hematopoietic elements. Its incidence is estimated to be 1: 280,000. There are two types: primary HLH is an autosomal recessive disorder affecting children most commonly within the first year of life. Secondary HLH is acquired in adults because of strong immunologic activation from systemic infection, autoimmune disease or underlying malignancy. The diagnostic criteria of fever, splenomegaly, cytopenia, hyperferritinemia, hypertriglyceridemia, soluble CD25 receptor level greater than 2400 pg/ml and hemophagocytes on bone marrow biopsy was developed for use in children and is likely less reliable in adults. The patient presented with 6/8 diagnostic criteria and a diagnosis of HLH was established. Although HLH has been reported as a sequela of Coxsackie Virus infection, the patient’s likely trigger was his lymphoma. Treatment involves controlling inflammation with corticosteroids and cyclosporin A. The use of etoposide and anti-thymocyte globulin removes activated immune cells. Treatment of underlying infections if present, supportive care with transfusions and prophylactic antimicrobials is also necessary.
Conclusions: This patient represents a rare case of HLH likely secondary to Non-Hodgkin Lymphoma. There is a paucity of data on the overall incidence of secondary HLH in the adult population. The entity should be considered in the differential diagnosis of patients with unexplained constitutional symptoms accompanied by cytopenia.