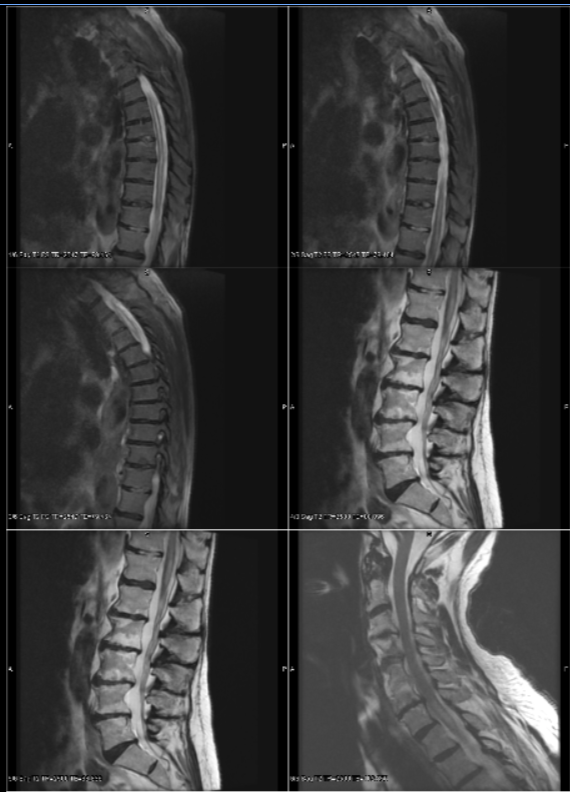
Case Presentation: A 77-year-old male with a history of lumbar spondylosis presented to the emergency department with 12 hours of lower extremity weakness, urinary retention, and saddle anesthesia. Two weeks before this visit, the patient presented to the emergency department with intractable hiccups, nausea, and vomiting and was ultimately discharged on baclofen and metoclopramide with his symptoms resolving one week later. During his most recent presentation, he had a mild postural tremor in the hands, severe weakness to bilateral hip flexion, and moderate weakness to knee extension and flexion and feet dorsiflexion. Deep tendon reflexes were normal and symmetric in the arms, 1/4 for the knees, and depressed at the ankles. Babinski reflex was indifferent bilaterally. He had severe pinprick sensation loss in the lower extremities and torso from T2 and inferiorly. A spine survey MRI showed a heterogeneous T2 intensity from C7 to the distal conus medullaris. These findings were pathognomonic for NMOSD. Lumbar puncture showed WBC of 350 with 90% neutrophils, RBC of 0, CSF protein of 238, and CSF glucose of 41 with a negative infectious workup. He was started immediately on high-dose intravenous methylprednisolone and plasma exchange. Emergent brain MRI showed T2 intensity involving the area postrema. Repeat thoracic and cervical spine MRI showed subtle patchy enhancement in the mid- and lower thoracic cord. Aquaporin-4 and Myelin-oligodendrocyte glycoprotein antibodies were sent out. Overnight, his paraparesis progressed to complete paraplegia with abrupt and complete sensation loss in the same distribution (T2 and inferior). He completed seven sessions of plasmapheresis combined with five days of IV high-dose steroids. He transitioned to a four-week prednisone taper with plans for long-term immunosuppression using Rituximab. On hospital day 19, AQP4 antibodies with titers >1:100000 confirmed the patient’s diagnosis of neuromyelitis optica spectrum disorder.
Discussion: NMOSD is an inflammatory condition of the CNS with devastating sequelae. High clinical suspicion can lead to early recognition and treatment which might improve outcomes. In this case, the initial presentation of unexplained hiccups, nausea, and vomiting was consistent with area postrema syndrome but went undiagnosed. This patient’s second presentation was consistent with severe acute myelitis which confirmed the diagnosis and prompted immediate and aggressive immunotherapy. Other notable presentations include acute vision loss from optic neuritis and excessive somnolence or narcolepsy from diencephalon involvement. The case demonstrates the high acuity and morbidity of this disease as the patient went from a state of normal functionality to paraplegia in one day. One might argue that early recognition of the area postrema syndrome would have led to earlier aggressive immunotherapy and potentially would have prevented the second presentation of severe myelitis associated with severe morbidity.
Conclusions: Neuromyelitis optica spectrum disorder (NMOSD) is a rare, autoimmune condition that results in severe demyelination and axonal damage of the central nervous system (CNS). Initially considered a subvariant of multiple sclerosis, NMOSD is recognized as a distinct disease with differing immunopathogenesis and treatments. Untreated, NMOSD carries a poor prognosis and high morbidity, thus early recognition and treatment are crucial.