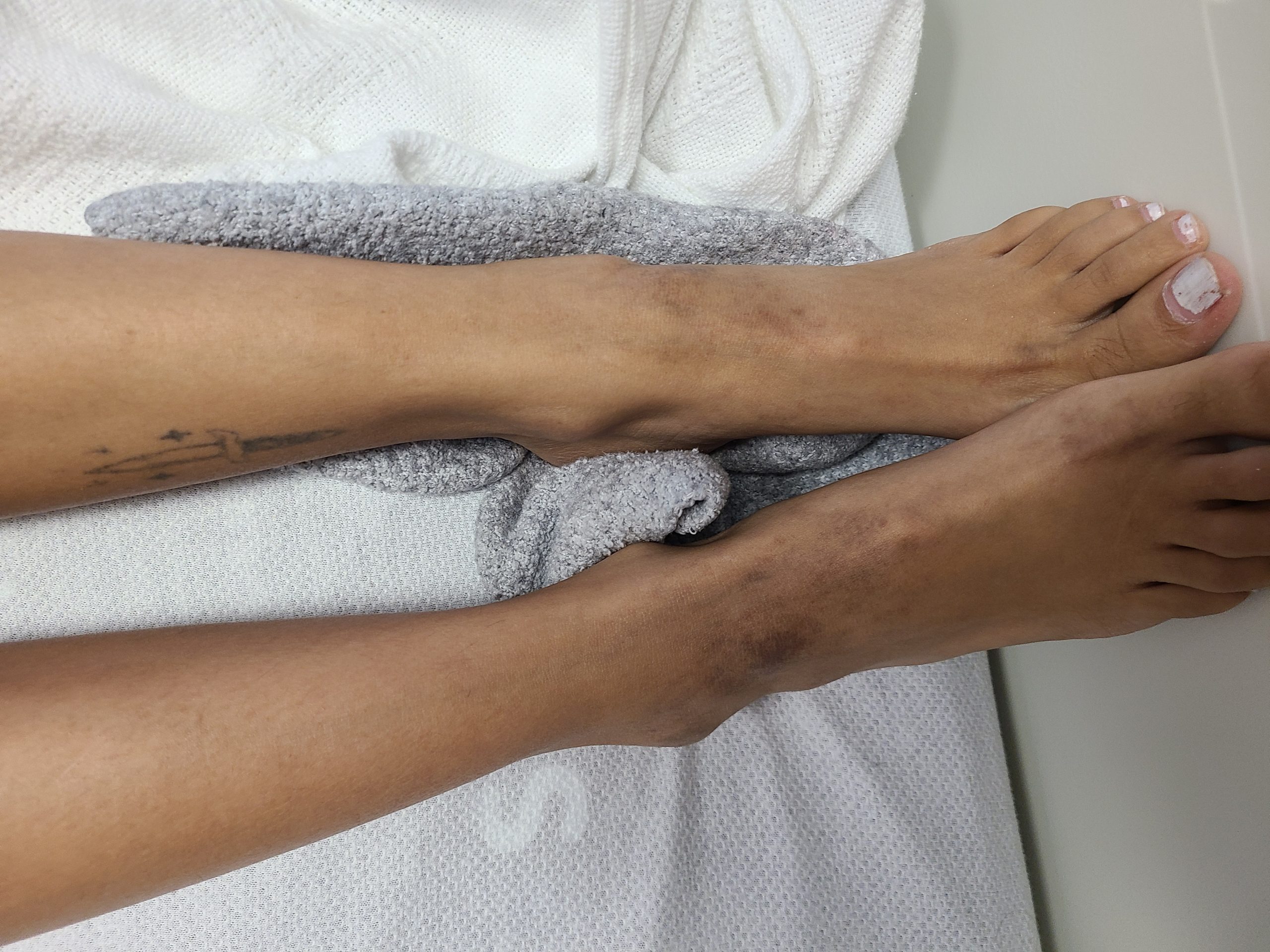
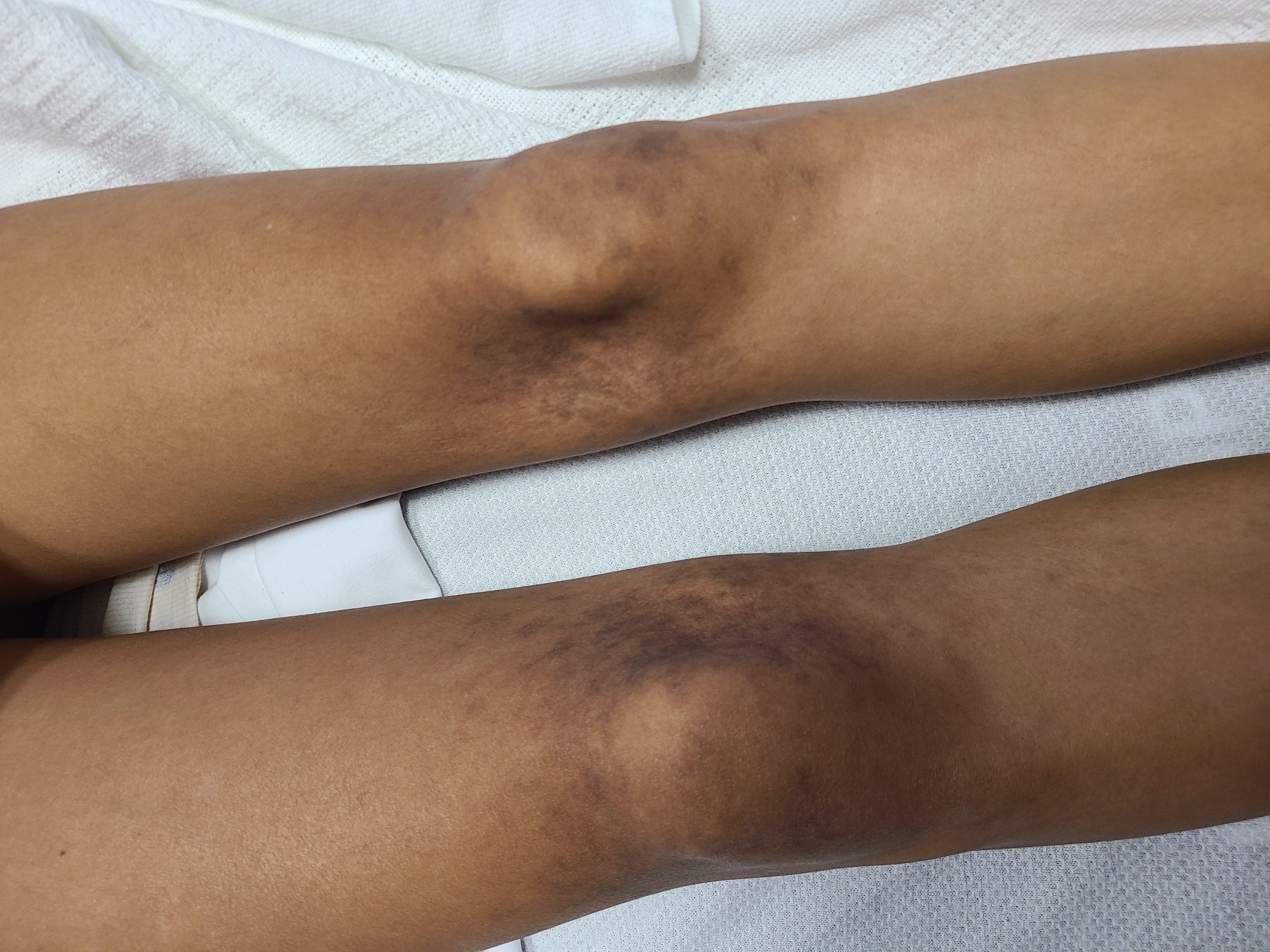
Case Presentation: A 19-year-old female with no significant past medical history presented to our center with recurrent unprovoked epistaxis episodes, gingival bleeding, a non-pruritic lower extremity rash, and fifteen-pound weight loss over three months. She reports associated episodes of headaches and dizziness; however, denied oral ulcers, joint swelling or pain, muscle pain or weakness, Raynaud’s phenomenon, eye symptoms, fever, abnormal bruising, recent travel or sick contact, or history of smoking. On the physical exam, she had stable vitals. The rest of the exam was significant for a BMI of 16.46 and non-palpable, symmetric rash on medial aspects of the knees and posterior thighs of a reticular and lacy pattern. Additionally, the fundus exam showed bilateral dot blot hemorrhages throughout the periphery. Lab work was significant for hypochromic microcytic anemia, anisocytosis, and marked rouleaux on peripheral smear, iron deficiency anemia, huge polyclonal albumin gap, elevated IgG, IgA, and IgM levels >6,400, blood viscosity of >6.34, rheumatoid factor of >100units, Anti-CCP >300U/mL, ANA >1:640, Smith/RNP of 3.4, CK of 1328, ACE of 95U/L, and ESR of 73. Her SPEP, bone marrow and lymph node biopsy, coagulation studies, and platelet function test were unremarkable. Extensive infectious workup was unremarkable. CT scan of the Chest, abdomen and pelvis showed prominent bilateral axillary lymph nodes ranging up to 13 x 12 mm. She continued to deny any musculoskeletal symptoms, but based on significant elevated aldolase and CK, an MRI of the thigh showed mild increased T2 signal intensity in the bilateral semimembranosus muscles. This was followed by a thigh muscle biopsy that showed severe necrotizing inflammatory myopathy manifesting as lymphocytic infiltrates in perivascular spaces of perimysial vessels and Type specific atrophy of type 2 myofibers. The extended myositis panel was positive for Ribonucleic protein IgG Antibody 52 units, specifically Fibrillarin (U3 RNP). The patient received plasma exchange twice for the hyperviscosity and was started on prednisone 40 mg daily with plan for taper. Her viscosity and CK levels normalized at the time of discharge.
Discussion: Hyperviscosity Syndrome (HVS) is a medical emergency characterized by bleeding, visual disturbances, and neurological deficits. [1] Differentials include hematological malignancies, infectious etiologies, and rheumatological disease. Polyclonal gammopathy argues against a hematological etiology and favors infectious or autoimmune causes. This case of HVS is unique due to elevated immunoglobulins as an early presentation of undifferentiated connective tissue disease (UCTD). There are several case reports in the literature of patients presenting with hyperviscosity due to RA, including a case report in UCTD [2]; however, these patients usually have a long-standing history with manifestations such as Felty’s Syndrome. [3]. The usual treatment of hyperviscosity syndrome focuses on symptomatic management with plasma exchange and treating the cause. [4] Since the patient was not meeting specific criteria for CTD like SLE and RA, the decision was made to start the patient on 1mg/kg of prednisone daily with tapering based on response.
Conclusions: Hyperviscosity is rare and presents in many difficult-to-diagnose disease processes. This can lead to delays in diagnosis in treatment. However, given its emergent nature, at times, prompt identification and initial therapy with plasma exchange is key to reducing patient morbidity.