Case Presentation:
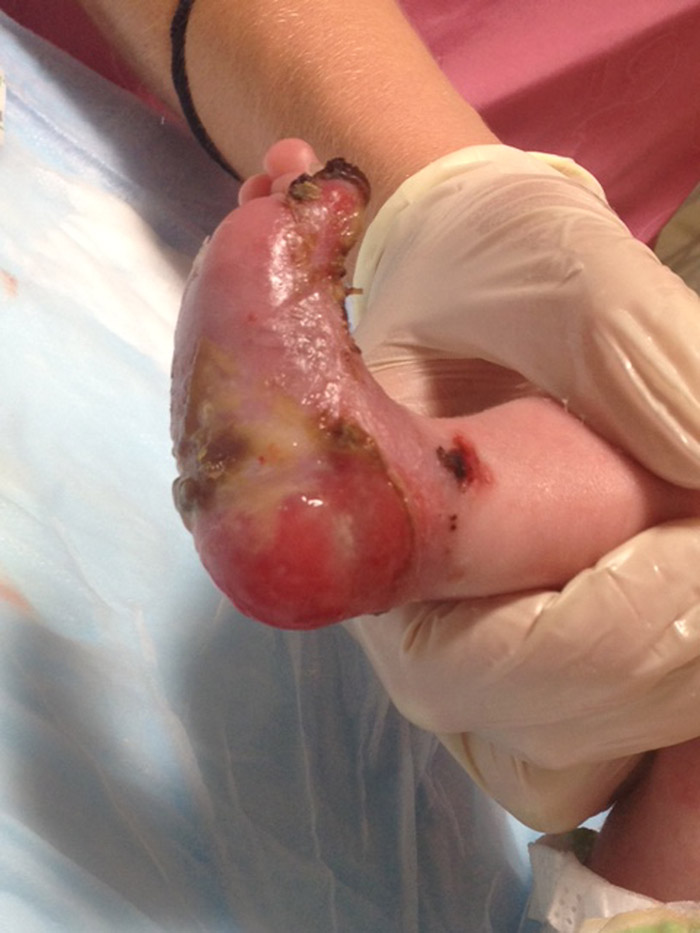
A 1‐day‐old infant male presented from a referring hospital with skin sloughing. He was born via caesarean section at 34 and 6/7 weeks estimated gestational age due to maternal HELLP syndrome. The pregnancy course was otherwise uncomplicated. His mother had no past medical history and took no medications during the pregnancy. Her serologies were unremarkable. At birth, the infant was noted to have diffuse blistering of the skin and epidermal loss of the lower extremities. Ampicillin, cefotaxime, and acyclovir were empirically started. A complete blood count, blood culture and serum herpes simplex virus polymerase chain reaction were negative. Upon transfer, he was afebrile with vital signs appropriate for his age. Physical exam revealed diffusely scattered bulla in various stages of evolution involving the face, chest, back, and extremities. Significant epidermal loss was noted over the plantar and dorsal surfaces of the bilateral lower extremities. A skin punch biopsy was performed and was sent for electron microscopy and immunohistochemistry. The results revealed significantly decreased collagen VII staining, consistent with dystrophic epidermolysis bullosa. Sequencing of the COL7A1gene, as recommended by Genetics consultants, confirmed the presence of two mutations and established the diagnosis of recessive dystrophic epidermolysis bullosa (RDEB). With the assistance of Dermatology and Plastic Surgery consults, wound care education was provided to the infant’s caregivers. He was ultimately discharged home in the care of his mother with close outpatient monitoring for signs of secondary infection and demonstration of adequate growth.
Discussion:
Although epidermolysis bullosa has been well reported, RDEB is a rare, severe subtype with an incidence of 0.4‐0.6 per million live births. It is caused by mutations within the COL7A1gene encoding type VII collagen, a protein that forms anchoring fibrils in dermoepidermal junctions. Blistering starts spontaneously at birth or after mild trauma. Occasionally, as in this case, there is extensive denudation of a body area due to congenital absence of the epidermis. Healing may occur with scarring and milia. RDEB carries an 8% mortality within the first year of life and there is significant morbidity associated. Infants, therefore, require specialized care through a multidisciplinary approach. Meticulous wound care is of the utmost importance, as infection is a known and deadly complication. Scar tissue that forms from healing wounds can cause severe contractures. Squamous Cell Carcinoma (SCC) is the most serious long‐term complication. By mid‐adulthood, nearly all will have at least one SCC, and nearly 80% will die of metastatic disease.
Conclusions:
RDEB, a serious congenital skin condition, is a rare subtype of epidermolysis bullosa that presents at birth and carries with it significant morbidity and mortality.