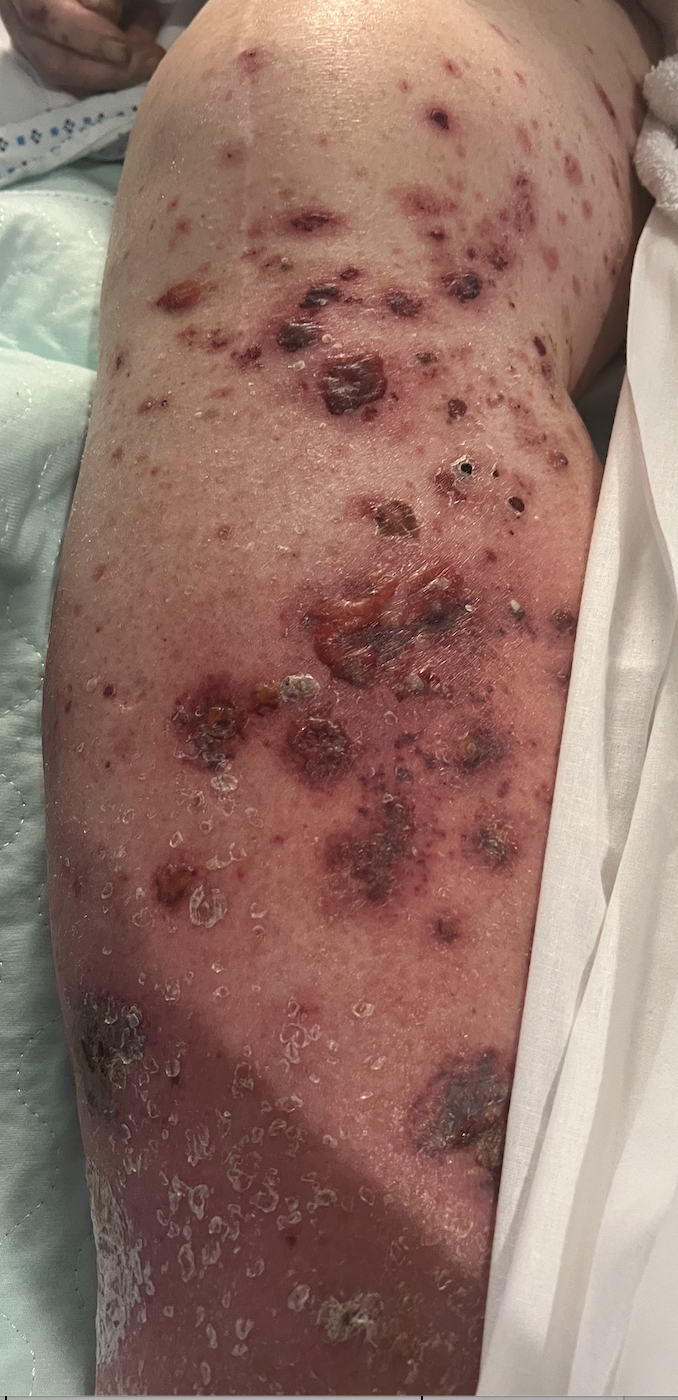
Case Presentation: An 83-year-old male with a history of seizures, CHF, hyperlipidemia, obstructive sleep apnea, and BPH presented with 2-3 days of altered mental status. His family reported worsening leg swelling and blisters for the past few months, with a rash spreading to his upper extremities and wounds draining serosanguinous fluid. He was hypoxic in the ED, with lab results showing elevated potassium, BUN, creatinine, and liver enzymes. Physical exam revealed pitting edema, hemorrhagic papules, and cellulitis. Chest X-ray indicated vascular congestion. He was started on broad-spectrum antibiotics for suspected cellulitis and DVT was confirmed via ultrasound. Dermatology noted significant skin changes suggestive of thromboembolic septic vasculitis, and a broad differential was considered, including lupus and vasculitis. A skin biopsy confirmed IgA vasculitis and nephropathy. Treatment included rituximab and steroids. His course was complicated by renal failure and pseudomonal bacteremia, leading to septic cardiomyopathy and cardiogenic shock. After 3 weeks of aggressive treatment, with ongoing deterioration on multiple vasopressors, the family opted for DNR status.
Discussion: The annual incidence of IgA vasculitis (IgAV) in adults is 0.8-2.2 cases per 100,000, with a male predominance (1.5:1 ratio). It’s associated with hypertension, diabetes, and obesity, stemming from IgA1-dominant immune complex deposition, triggered by mucosal antigens in genetically predisposed individuals. Although rare, links to malignancies exist, particularly solid tumors. HLA-DR1*0103, is strongly associated with susceptibility, though its ethnic implications are unclear.In adults, IgAV presents with symptoms like cutaneous purpura, arthralgia (61%), enteritis (48-53%), and glomerulonephritis (45-85%). Severe cases show necrotic and bullous lesions, with renal involvement being a key factor in morbidity and mortality. Treatment for mild cases is symptomatic, while severe manifestations require immunosuppressive therapy. Cyclophosphamide is often used, though guidelines for adults are lacking. Rituximab’s role remains debated, and long-term outcomes are challenging to predict due to limited studies.Classic symptoms include abdominal pain, purpura, and arthralgia, often related to bowel ischemia. Complications like intussusception require surgery. While pediatric protocols suggest prednisone for abdominal symptoms, there’s no standardized adult treatment. Our patient, treated with Rituximab and steroids, faced complications including renal failure and hypotension. An multidisciplinary approach is crucial for effective management of IgAV, highlighting the need for early diagnosis and intervention to prevent severe outcomes.
Conclusions: Henoch-Schönlein purpura (HSP) is self-limiting, with most patients recovering within 8 to 10 weeks; however, about 5% may develop chronic symptoms. Full resolution is more likely in cases with mild renal involvement and no neurological issues, particularly if the disease duration is under 6 weeks. Recurrences occur in 30% to 50% of patients, and long-term studies indicate a risk of CKD, especially with steroid treatment. The absence of established treatment guidelines complicates management, necessitating a balance between disease control and treatment risks. Research into biologics for severe IgAV is essential, as early use may help reduce toxicity. Clinicians should be cautious with immunosuppression affecting survival.