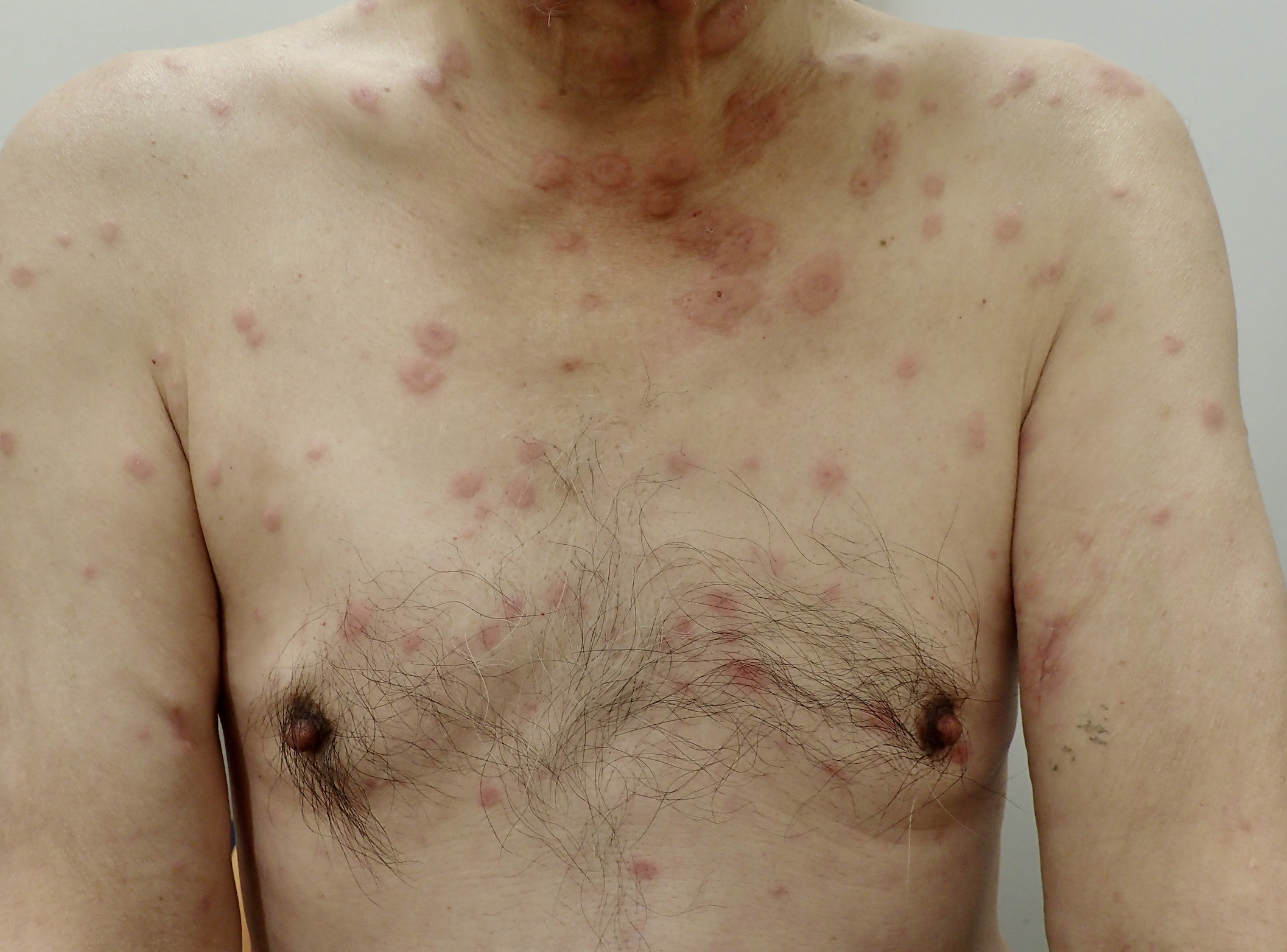
Case Presentation: A 70-year-old man visited our hospital for lower extremity edema and involuntary weight loss. A routine check-up at his diabetes clinic five months earlier revealed anemia and an increased inflammatory response. His medical history included hypertension, dyslipidemia, and type 2 diabetes. Computed tomography (CT), blood cultures, gastrointestinal endoscopy, and colonoscopy findings were unremarkable; however, his symptoms persisted, leading to a referral to our hospital. A physical examination showed normal vital signs and pitting edema in the extremities, without joint tenderness or skin rash. Laboratory tests showed normochromic anemia and elevated C-reactive protein levels. Test for rheumatoid factor, antinuclear antibody, antineutrophil cytoplasmic antibody, and anti-SS-A antibody were negative. Contrast CT findings were unremarkable. Bone marrow aspiration (BMA) indicated hypercellular marrow with mild erythroid dysplasia. Fluorine-18-fluorodeoxyglucose positron-emission tomography showed a slightly high uptake in the spleen and BM. After consultation with a rheumatologist and hematologist, a tentative diagnosis of remitting seronegative symmetrical synovitis with pitting edema syndrome was made. The patient was started on prednisone 20 mg/day. Initially, his condition improved but it soon deteriorated with erythematous, edematous plaques appearing all over his body (Figure 1), requiring hospitalization. Skin biopsy revealed atypical lymphoid and neutrophilic infiltration in the dermis; however, immunostaining and flow cytometry did not identify abnormal lymphocytes. Although the final diagnosis remained elusive, an increased prednisone dose of 60 mg/day alleviated the symptoms and led to his discharge. Given the patient’s unresolved dermatitis and hematological abnormalities, VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome was suspected. A careful review of the BM examination revealed vacuoles in granulocytes and erythroblasts (Figure 2). Genetic testing confirmed the presence of somatic UBA1 variant (p.Met41Leu), resulting in a VEXAS syndrome diagnosis.
Discussion: VEXAS syndrome predominantly affects older men and is characterized by UBA1 somatic variants, inflammatory manifestations, hematological abnormalities, and vacuoles in myeloid and erythroid precursors. Its primary clinical features are skin lesions (83%), fever (64%), weight loss (62%), lymphadenopathy (34%), and arthralgia (27%). Skin symptoms are common and diverse, ranging from red or violaceous papules to inflammatory edematous papules, erythematous plaques, and livedo racemosa. Nearly all affected patients show vacuoles in myeloid and erythroid progenitors. Furthermore, myelodysplastic syndromes occur frequently in affected patients, with incidences of 25%–55%. High-dose glucocorticoids remain the only effective treatment for VEXAS syndrome, although long-term use often leads to resistance. Alternative effective treatments remain unidentified.
Conclusions: We described a case of VEXAS syndrome characterized by persistent edema and weight loss. Initially, the diagnosis was challenging owing to the absence of cutaneous symptoms and the presence of inconspicuous vacuoles in the BM. In cases of persistent dermatitis of an unknown cause, BMA can contribute to the diagnosis of VEXAS syndrome. VEXAS syndrome must be considered in older men presenting with unexplained inflammation and a transient glucocorticoid response.