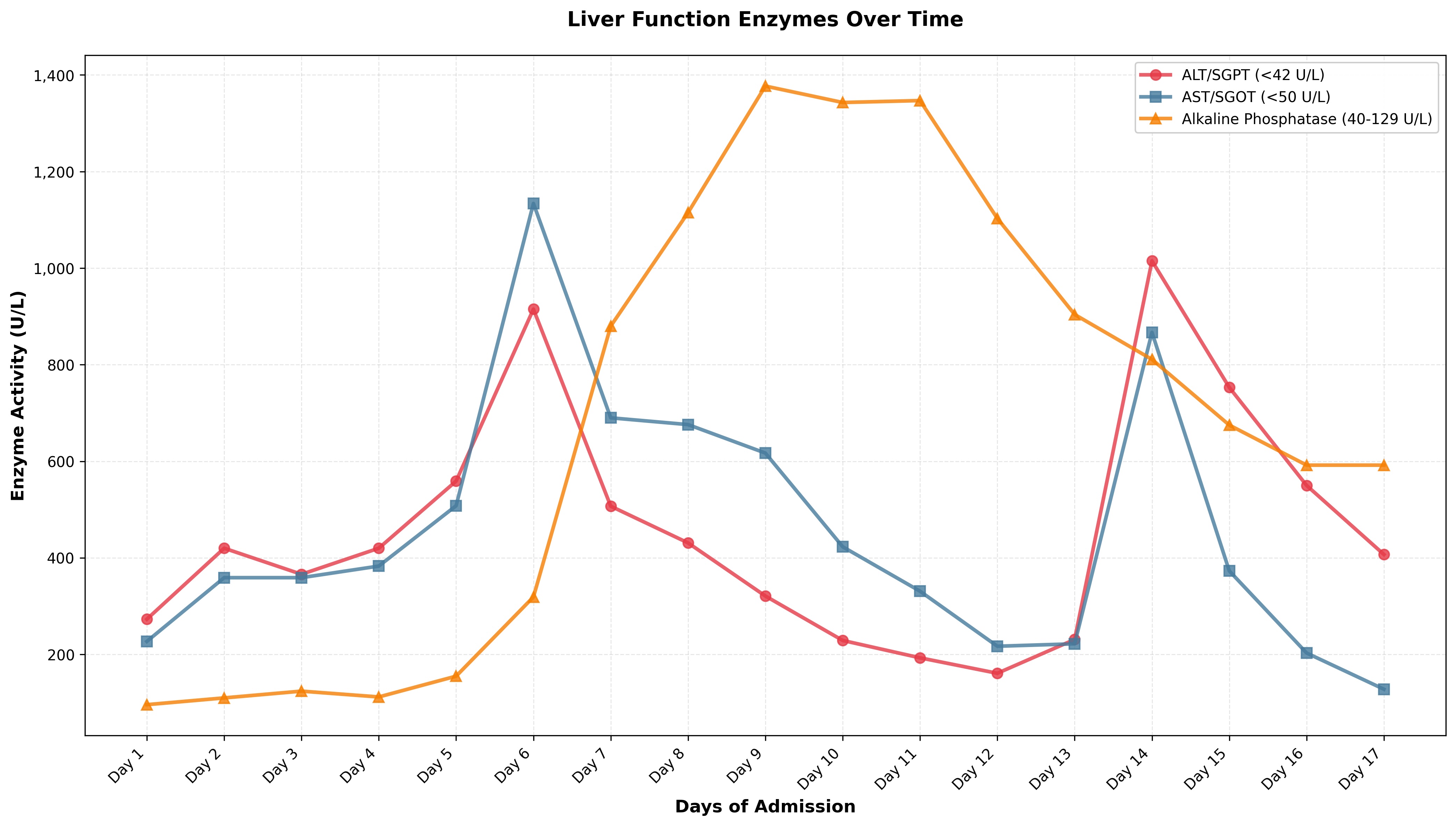
Case Presentation: We present a 25-year-old African American male with a known history of Hb SC disease who presented with fevers up to 104°F and an elevated ferritin of 22k with AST 179/ALT 235 and ALP of 110. Full infectious workup was negative. Hemoglobinopathy panel confirmed the patient was in acute sickle cell crisis, and an exchange transfusion was completed; he was on pain medications.Over the next 6 days, his ferritin increased to 81k, with AST 1134 and ALT up to 915. His hemoglobin declined below 7 g/dL, requiring transfusion of 3 units of PRBCs, and his platelet count dropped to 38,000/µL, for which he received 2 units of platelets. CT abdomen showed mild splenomegaly, >2 cm below the costal margin. Fibrinogen was 301, Triglycerides were 155, and Soluble CD2 was elevated up to 2,496. Liver biopsy showed mild to moderate mononuclear lobular inflammation without necrosis or steatosis, Kupffer cell hyperplasia with rare hemophagocytosis. The sinusoids exhibit mild congestion with sickled red blood cells but without dilation. Foci of hepato-canalicular cholestasis and rare acidophil bodies were noted. No viral inclusions identified. Peripheral flow cytometry did not show any evidence of lymphoproliferative diseases. Liver biopsy showed a high iron index. Given that he met several HLH criteria, corticosteroids were started.His hospital course was complicated by new-onset acute hypoxic respiratory failure, which was likely due to HLH. He also had a small BL PE for which anticoagulants were administered. He developed severe abdominal pain a few days after the liver biopsy and was found to have hepatic hematoma with a negative hepatic angiogram, and his anticoagulation was stopped. Both complications had overlapping features with sickle cell pain crisis/ sickle cell hepatopathy.
Discussion: The patient’s history of sickle cell disease and concern for hemophagocytic lymphohistiocytosis (HLH) were concurrent. The presence of rare hemophagocytosis can support the clinical impression of HLH but should be interpreted alongside other criteria, as it can occur in patients with sickle cell disease. He had sickled RBCs seen within non-dilated sinusoids; however, the absence of biliary obstruction and lack of necrosis made sickle cell hepatopathy less likely. HLH mimics sickle-cell complications, as features of anemia, pain, high ferritin, and organ dysfunction overlap. This makes the diagnosis very nonspecific. Sickle cell patients may have hyperferritinemia due to iron load from multiple transfusions compounded by acute inflammatory illnesses (1). The clinical pathway to diagnose HLH is confirmation of 5/ 8 diagnostic criteria, including fever, bicytopenia, splenomegaly, hyperferritinemia, hypertriglyceridemia or hypofibrinogenemia, hemophagocytosis, elevated soluble interleukin-2 receptor, and low/absent natural killer (NK)-cell activity (2). Most cases of HLH in sickle cell disease in the literature were treated with supportive care, including Immunosuppressants, steroids.
Conclusions: HLH presents a diagnostic challenge because there is no one pathognomonic clinical manifestation or laboratory finding, and signs are often nonspecific(3). Given the aggressive nature of HLH, prompt recognition and initiation of treatment are key. This case illustrates the importance of having a high suspicion for HLH in patients with sickle cell anemia with fever, pancytopenia, and multi-organ dysfunction.
.jpg)