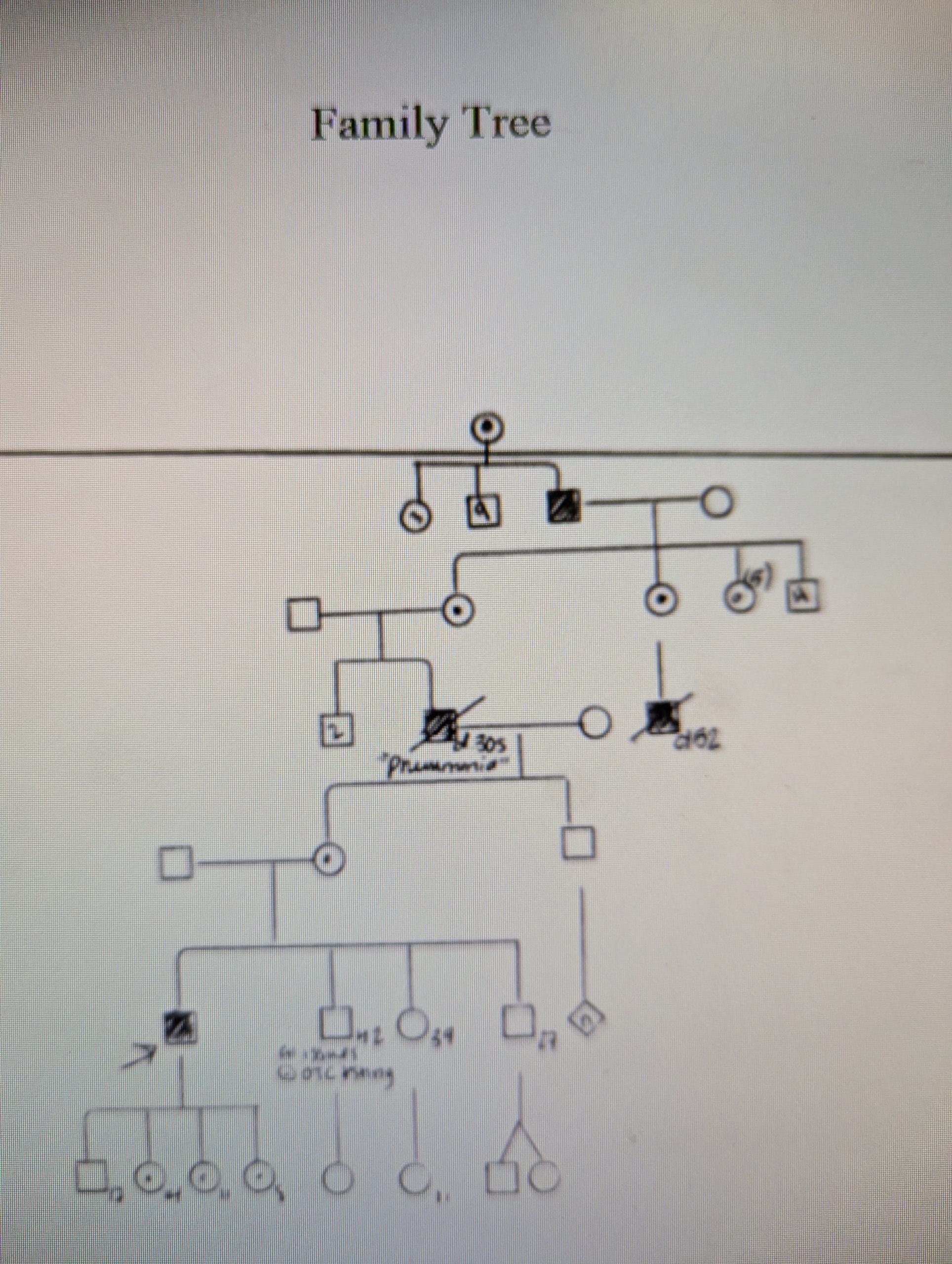
Case Presentation: 44-year-old male with a history of seasonal allergies presented with subacute mental status and behavior changes, malaise, and decreased oral intake after elective nasal polypectomy. Following polypectomy, he was started on a nasal steroid and antibiotics. Local ED determined symptoms possibly due to recent surgery. IVF and PO glucose provided temporary symptom relief. Family history notable for unexplained male deaths at varying ages on maternal side with mention of post-mortem diagnoses of a genetic condition. On presentation he is A&O x3, afebrile, hemodynamically stable with no neurologic deficits; his behavior since discharge from local ED was odd, including attempting to utilize a tissue box as an ATM machine. LFTs, CBC, and BMP were unremarkable; the only notable lab was an elevated ammonia. UDS was negative and UA notable for 1+ ketones. CT brain, CT maxillofacial/sinus and MRI brain w/wo were unremarkable. Continuous D5-NS and lactulose BID were started. After genetics consult given family history, lactulose was discontinued, lipid infusion initiated and fluids adjusted to D10-NS+10KCl in attempt to suppress catabolism with concern for underlying urea cycle disorder. Ammonia levels jumped from 89 to 228 in 4 hours and peaked at 292 without mental status or neurologic changes. For his rapidly increasing ammonia, a central line was placed in preparation for nitrogen waste scavenger treatment. His course was complicated by two days of mania vs psychosis characterized by grandiose thoughts, pressured speech and visual/auditory hallucinations. These were believed secondary to retained ammonia in his CNS, and resolved with continued catabolism suppression. Fluids were discontinued, and he was started on a low protein diet. Subsequent ammonia levels x 24 hours normalized prior to discharge. After discharge, plasma and urine amino acids and urine orotic acid results returned to confirm the diagnosis of OTC deficiency. His family’s rare OTC c.-106C>A variant was detected on further genetic testing. He is being treated with citrulline and arginine supplementation and follows a low protein diet to help avoid future hyperammonemic crises.
Discussion: OTC deficiency is the only X-linked urea cycle disorder and has varying degrees of severity. Although it is X-linked females may develop this disease due to mosaicism in hepatocytes. Prevalence is estimated at 1-9/100,000, with < 20% presenting after age 6, and even fewer after age 18. Adult onset OTC deficiency usually presents emergently to the inpatient setting with hyperammonemic crisis secondary to environmental stressors leading to a catabolic state. For this patient, these likely were a combination of surgery, fasting, and steroid use. Hyperammonemia is a common in hospital problem and inpatient lab abnormality with varying etiologies and frequently presents as non-specific altered mentation and behavior changes for which admission and thorough workup and treatment are necessary.
Conclusions: A thorough family history is critical in identifying rare metabolic disorders, because these diseases are rare and the presentation of hyper ammonia is non-specific; the differential diagnosis for hyperammonemia is broad and should include non-liver disease related conditions. As exemplified in this family’s history of repeated unexplained male deaths in his maternal side, OTC deficiency is difficult to diagnose and delays in identifying urea cycle disorders and other metabolic conditions can cause permanent neurologic damage or death.