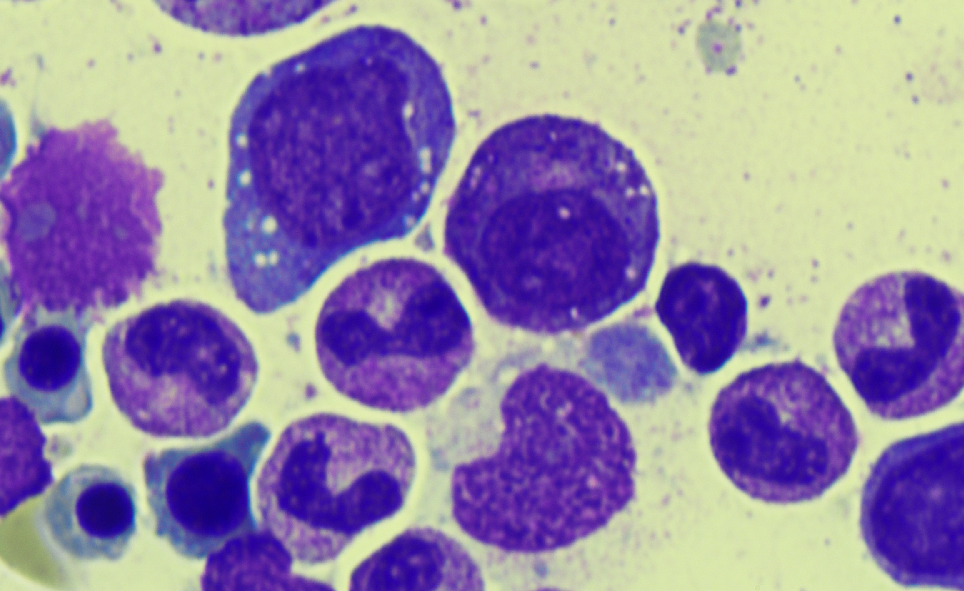
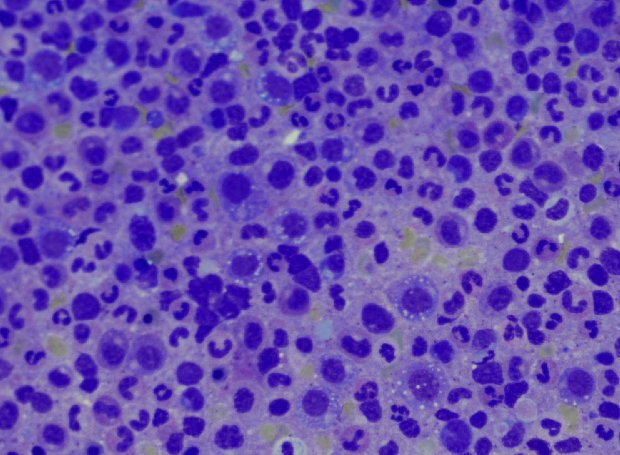
Case Presentation: A 76 year old male without any significant past medical history presented to the emergency department with abdominal pain, night sweats, and a fever for a week. He was found to have a mild macrocytic anemia (Hgb 9.9, MCV 105.4) with a normal B12 and folate, and elevated inflammatory markers (CRP 24, ESR 55, ferritin 1,321). A CT revealed significant abdominal ascites, mesenteric edema, and hepatic surface nodularity. A liver biopsy showed necroinflammatory injury and reticuloendothelial iron without evidence of chronic liver disease. Ascites fluid contained 576 neutrophils without any organism identified and a SAAG < 1.1. Over the course of the hospitalization the initial presentation of ascites and abdominal pain were complicated by the presentation of new findings including pulmonary emboli, a persistent cough, migratory polyarthralgias, a non-blanching erythematous rash on the upper thighs and chest, a worsening macrocytic anemia (Hgb 6.4), and thrombocytopenia. Skin biopsy revealed a neutrophilic dermatosis. A bone marrow biopsy was completed and demonstrated multiple vacuoles in early erythroid and myeloid precursors. The patient’s unique constellation of symptoms and specific bone marrow finding were suspicious for the inflammatory syndrome VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome). Given high clinical suspicion the patient was started on methylprednisolone 1mg/kg/day and had a remarkable improvement in symptoms including resolution of arthralgias, abdominal pain, and night sweats.
Discussion: VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome) is a treatment-refractory inflammatory syndrome associated with hematologic abnormalities first described in 25 patients in 2020. The syndrome is due to a nonsense somatic mutation in the UBA1 gene, encoding for the major enzyme that initiates ubiquitylation, in hematopoietic bone marrow precursors. Clinically, the syndrome has been identified in men ages 50-70 many of whom present with some combination of constitutional symptoms, vacuoles in erythroid and myeloid cells on bone marrow biopsy, macrocytosis, thrombocytopenia, elevated inflammatory markers, ear and nose chondritis, vasculitis, arthralgias, a variety of rashes, thromboembolisms, hepatosplenomegaly, and pulmonary involvement (infiltrates, effusions). Treatment for the syndrome has largely consisted of high dose steroids. To our knowledge there have been no reports of abdominal ascites in a patient with VEXAS, making this the first report of an alternative inflammatory symptom.
Conclusions: VEXAS is a newly described late-onset somatic mutation leading to a host of inflammatory and hematologic abnormalities that are continuing to be discovered as we learn more about this syndrome and its true prevalence. Our case represents the first report of ascites as a component of the VEXAS associated inflammatory symptoms, revealing the importance for hospitalists to consider VEXAS on the differential diagnosis for ascites without a clear etiology.