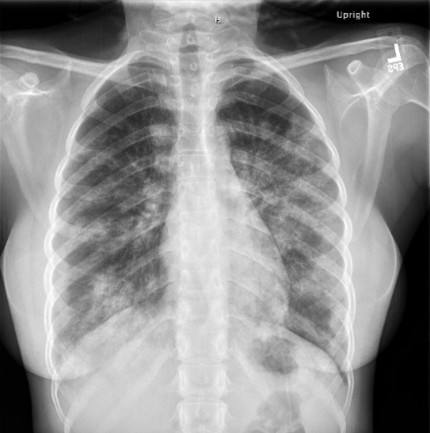
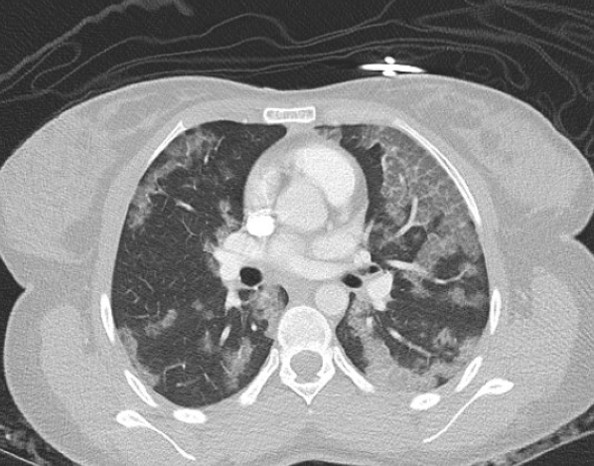
Case Presentation: A 17-year-old girl presented with one month of progressive cough and exertional dyspnea. She denied fever, chest pain, or hemoptysis. Vitals were normal for age. Growth review showed recent weight loss from the 38th to 15th percentile with stable height at the 5th percentile. Pulmonary exam revealed faint bilateral crackles but was otherwise unremarkable. Chest radiograph demonstrated diffuse bilateral air-space consolidations. CT angiogram revealed extensive ground-glass opacities and interstitial thickening. Laboratory studies (CBC, CMP, ESR, CRP) were unremarkable. Empiric antibiotics for presumed atypical pneumonia were started on admission but soon discontinued when her course appeared inconsistent with acute infection. Pulmonology team consulted and recommended bronchoscopy with bronchoalveolar lavage (BAL). Bronchoscopy showed normal mucosa with mild friability and scant mucus. After BAL, the differential included infection, interstitial lung disease, and pulmonary alveolar proteinosis (PAP). The patient remained clinically stable without oxygen requirement and was discharged pending cytology results. BAL cytology revealed foamy, lipid-laden macrophages containing periodic-acid–Schiff (PAS)-positive granular cytoplasm and extracellular proteinaceous globules, confirming PAP. Video-assisted thoracoscopic biopsy later demonstrated anti–GM-CSF-antibody–negative disease, consistent with non-autoimmune PAP.
Discussion: PAP is a rare disorder characterized by impaired surfactant clearance and intra-alveolar accumulation of lipoproteinaceous material. It may be congenital, autoimmune, or secondary to environmental, infectious, or malignant processes. Classic imaging shows bilateral ground-glass opacities; BAL typically demonstrates PAS-positive material. Autoimmune PAP is defined by circulating anti-GM-CSF antibodies, while congenital and secondary forms are antibody-negative. This case illustrates the diagnostic challenge of distinguishing PAP from more common causes of subacute or chronic cough in adolescents, such as atypical pneumonia or hypersensitivity pneumonitis. Anchoring bias toward infection could have delayed recognition. A systematic approach – including reconsidering the differential when initial therapy fails, early specialty consultation, and targeted diagnostics such as BAL cytology – facilitated accurate diagnosis. Hospitalists play a key role in identifying symptoms that are not consistent with common concerns and indications for admission.
Conclusions: When adolescents present with persistent respiratory symptoms and incongruent infectious findings, clinicians should broaden the differential to include several alternative underlying etiologies including interstitial lung disease, inflammatory pulmonary conditions, and surfactant-clearance disorders such as PAP. Maintaining diagnostic openness, avoiding premature closure, and engaging consultants early can expedite diagnosis and prevent unnecessary treatment.