Case Presentation: A 30 year old woman with history of benign essential hypertension, hyperlipidemia, gastroesophageal reflux disease and fatty liver disease who was four days post-partum after a vaginal delivery complicated by post-partum depression presented with acute onset altered mentation, erratic behavior, visual hallucinations, combativeness and occasional bursts of hyperactivity. She had been started on sertraline the morning of symptom onset. Due to her confusion, review of systems was obtained from her parents and included blurry vision, diarrhea, tremors and depression, in addition to what was listed above. Her home medication list included prenatal vitamin daily, metoprolol succinate daily, lansoprazole daily, sertraline daily and promethazine daily as needed. Her vital signs were within normal limits and her physical exam was significant for depressed mood, flat affect and orientation to only person and place. Initial laboratory data was significant for an ammonia level of 175umol/L, potassium of 3.2mmol/L, chloride 19mmol/L, bicarbonate of 19mmol/L, BUN of 5mg/dL and creatinine 0.39mg/dL. Anion gap, CBC with differential, PT/INR/PTT, folate, vitamin B12, TSH, acute hepatitis panel and liver function tests that were within normal limits. Fractionated serum amino acid testing was completed showing low levels of citrulline, taurine, valine and leucine. A liver ultrasound showed an unremarkable liver and normal hepatic vessels. Liver biopsy electron microscopy suggested microvesicular (mitochondrial) fat which is seen in fatty liver of pregnancy as well as urea cycle deficiency. Subsequent DNA sequencing was completed and helped confirm heterozygous urea cycle deficiency (UCD) and carbamoyl-phosphate synthetase (CPS1) deficiency. She was acutely started on lactulose and rifaximin with resultant improvement in her mentation. Long-term management was with a 50 gram protein diet, L-citrulline supplement and glycerol phenylbutyrate.
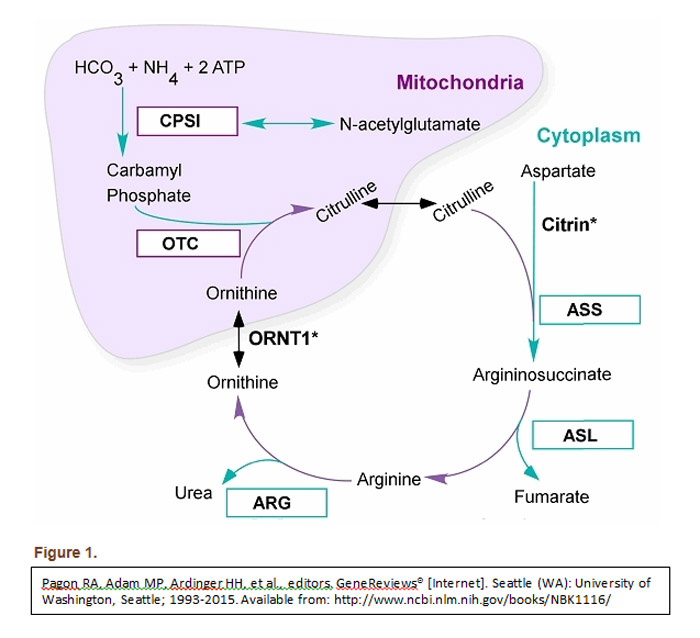
Discussion: This case highlights the rare condition of UCD and its variant CPS1 deficiency diagnosed in adulthood. The urea cycle is the liver’s primary mechanism for clearance of protein metabolism waste products (see figure 1). Data published by Summar et al in 2013 found the incidence of CPS1 deficiency to be an estimated 1:1,300,000. Case studies have reported that UCD can be trigged by childbirth. It is postulated that stressful physiologic states such as pregnancy trigger catabolism demanding increased hepatic metabolization of ammonia. Given the patient’s CPS1 deficiency, this quickly led to the liver’s maximum processing threshold being reached and hence her hyperammonemic state.
Conclusions: Urea Cycle Deficiency is typically overlooked given it rarity. Given the severe morbidity and high rate of mortality associated with UCD in adults, it must be considered in patients with otherwise unexplained hyperammonemia and hepatic encephalopathy.