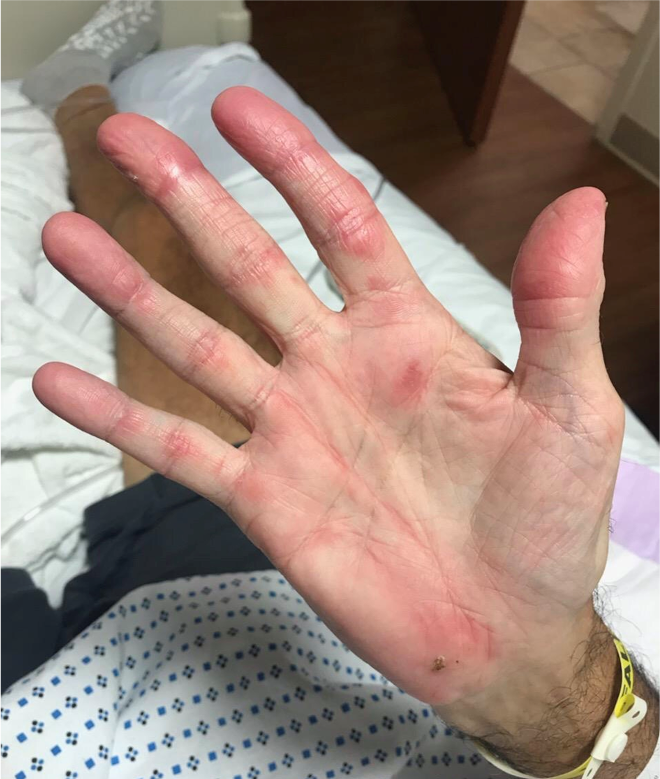
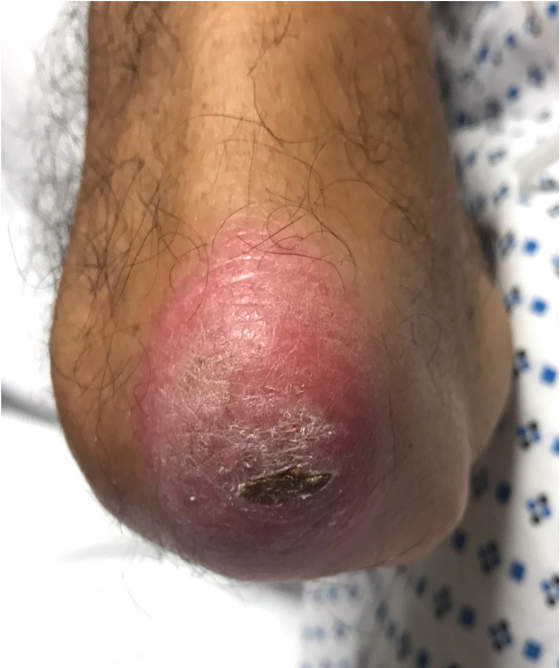
Case Presentation: A 68-year-old man with hypertension and osteoarthritis presented with fever, cough, dyspnea, and weight loss for 2 months. Prior to admission, he had received antibiotics for community-acquired pneumonia without improvement. Physical exam showed multiple ulcers at the base and sides of tongue, bilateral palmar erythematous papules with ulceration and scaling, bilateral erythematous plaques over the metacarpal, proximal and distal interphalangeal joints, tenderness over bilateral knees without synovitis, bibasilar crackles, and muscle strength was intact throughout. CT chest showed diffuse bronchiectasis with scattered ground glass and nodular opacities. Skin biopsy was suggestive of dermatomyositis showing interface dermatitis, however muscle enzymes were normal. A myositis panel revealed an elevated anti-MDA5 (melanoma differentiation-associated gene 5) antibody (>150) and an elevated ferritin level. Lung biopsy showed reactive inflammation and diffuse alveolar damage. Infectious and malignancy workup were negative. The patient was diagnosed with clinically amyopathic dermatomyositis (CADM) with rapidly progressive interstitial lung disease (RPILD). The patient developed acute respiratory distress requiring intubation despite treatment with pulse and high dose corticosteroids. He was treated with immunosuppressant agents including intravenous cyclophosphamide, tofacitinib, tacrolimus, and several courses of plasma exchange. Hospital course was complicated by acute renal failure requiring dialysis, bilateral pneumothoraces, inability to wean off mechanical ventilation, and septic shock due to bacteremia. The patient expired soon afterwards.
Discussion: Idiopathic inflammatory myopathies consist of autoimmune diseases that are characterized by muscle inflammation and include the commonly known dermatomyositis (DM), polymyositis (PM), and inclusion body myositis. The group also includes rare subtypes such as anti-synthase syndrome and CADM. DM is associated with pathognomonic skin findings (Gottrons’s papules, heliotrope rash), elevated muscle enzymes, and symmetric proximal weakness. Unlike classical DM, CADM patients can present without proximal weakness, normal muscle enzyme levels, cutaneous and digital tip ulcers, and are at a higher risk for developing RPILD. The atypical presentations often lead to delay in diagnosis and treatment. CADM patients with positive MDA5 antibody have worse prognosis than those without and anti-MDA5 levels correlate with the severity of skin ulceration and severity of RPILD [2,3]. Elevated ferritin levels are associated with poor prognosis [2]. Like DM, CADM can be associated with malignancies and therefore patients should be screened. Currently, there are no official guidelines for treatment of CADM. IV corticosteroids and other immunosuppressive agents (cyclophosphamide, tacrolimus, mycophenolate, cyclosporine) have been used, with some evidence showing that combined immunosuppressive therapy plus corticosteroid leads to a better survival rate [4]. Tofacitinib is a janus kinase inhibitor, which has been shown to improve survival and CT chest findings in a small group of patients [5].
Conclusions: Anti-MDA5- associated CADM is a rare subtype of DM, and can have an aggressive course with distinct cutaneous findings and RPILD. Management can be challenging. If suspicion is high, intravenous corticosteroids as well as immunosuppressant and biologic agents should be considered as early as possible.