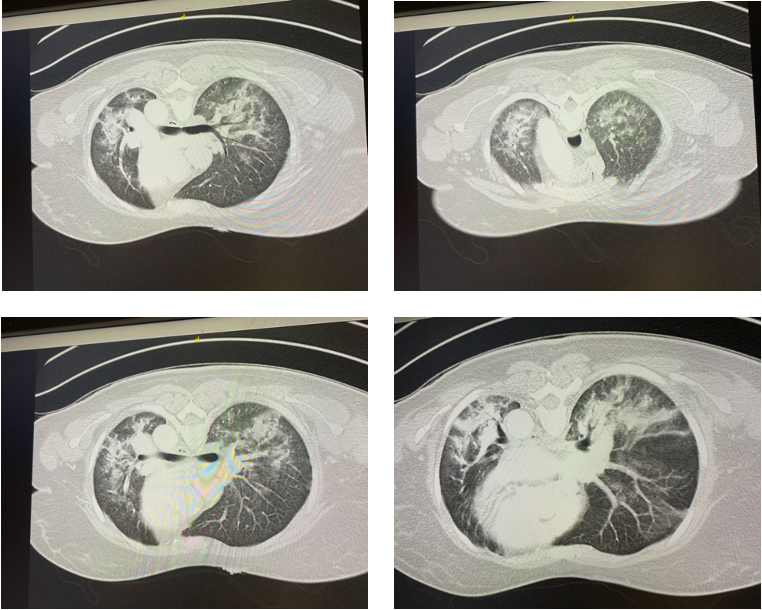
Case Presentation: A 40-year-old woman with no prior past medical history presented to the emergency room with several weeks of dysphagia, rash on her face, hands, chest, accompanied by fatigue, malaise, followed by progressively worsening shortness of breath. Over 8 weeks, she also developed a rash over her dorsal hands and upper chest wall, associated with dyspnea on exertion and dysphagia. She had no significant family history. She did not smoke cigarettes and did not consume alcohol. On exam, her vital signs showed a temperature of 101 degrees Fahrenheit, HR 133 beats/minute, respiratory rate 32 breaths per minute, O2 saturation 96% on 15 liters per minute of supplemental O2. On exam, she is a young woman in mild respiratory distress. Cardiac exam showed tachycardia without murmurs, rubs, or gallops. She spoke with 3 sentence dyspnea, and lung auscultation showed bibasilar rales with wheezes. Skin exam showed faint blanching erythema to the anterior chest wall, scaly erythema overlying the right 2nd and 3rd MCPs, and on the left 3rd MCP. Admission chest x-ray and CT of the chest showed multifocal opacities, most prominent in the lung bases, with prominent interstitial markings concerning for atypical pneumonia. Her respiratory status declined rapidly within 48 hours, and she required intubation. Due to worsening hypoxemia, she required extracorporeal membrane oxygenation (ECMO). A myositis-specific panel showed an elevated anti-melanoma differentiation-associated protein (MDA)-5 antibody. Despite empiric treatment with steroids, IV immunoglobulin, and broad-spectrum antibiotics, she failed to improve and was transitioned to comfort care measures. She passed away from rapidly progressive interstitial lung disease associated with clinically amyopathic dermatomyositis (CADM).
Discussion: Clinically amyopathic dermatomyositis (CADM) is a subset of dermatomyositis and is characterized by skin findings (tender palmar papules with deep ulcerations) without muscle weakness. One sub-type of dermatomyositis associated with anti-MDA5 positivity, is commonly seen in certain ethnic groups (Asians). Severe cases progress to acute lung injury with hypoxemic respiratory failure, and a hyperferritinemic syndrome resembling hemophagocytic lymphohistiocytosis. Treatment is with immunosuppression, including steroids, mycophenolate mofetil, azathioprine, IV immunoglobulin, or IV cyclophosphamide. There is a role for advanced therapies, such as extracorporeal membrane oxygenation and lung transplant in certain patients.
Conclusions: This highlights a case of fatal, rapidly-progressive interstitial lung disease from an inflammatory myopathy. While this is a rare cause of dyspnea, it is more common in patients with underlying inflammatory myopathies, specifically subsets of dermatomyositis. Clinicians should consider testing for myositis specific syndromes in this patient population. Rapid recognition can lead to earlier treatment, preventing a “last gasp” diagnosis.