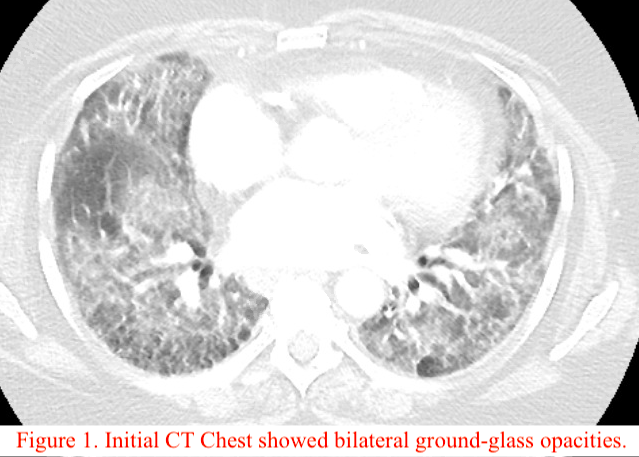
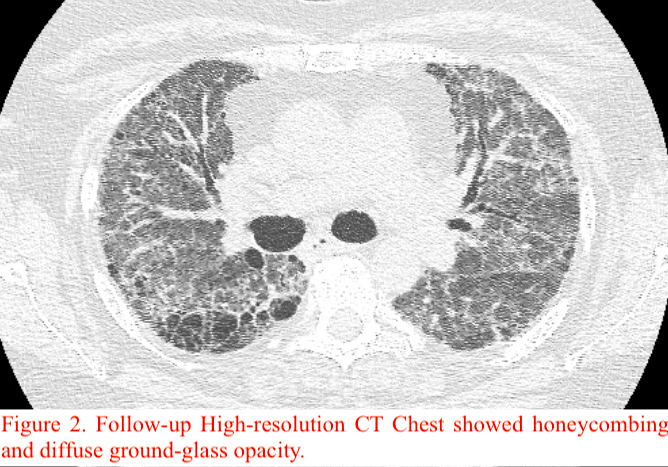
Case Presentation: A 61-year-old woman with past medical history of coronary artery disease, hypertension, hyperlipidemia, and a 46 pack-year history of smoking presented with gradually worsening shortness of breath and cough with white sputum production.The patient had multiple admissions in the last three months with similar presentation. In the first visit, CT chest showed bilateral ground-glass opacities (Figure 1) and bronchoscopy revealed 29% eosinophils on bronchoalveolar lavage, raising concerns for eosinophilic pneumonia presumed secondary to Hydrochlorothiazide and Wellbutrin. These were discontinued and the patient was discharged on oxygen. On the following admission, there was concern for rheumatoid lung given her positive RF (844) and anti-CCP (>200) titers. She improved with pulse dose steroids, and mycophenolate was started at discharge with the goal of tapering prednisone. On her next admission, the patient had acute hypoxic respiratory failure with CT findings of extensive honeycombing and diffuse ground glass opacity (Figure 2). An ILD specialist was consulted, and given her rheumatoid titers and chest imaging findings, the etiology was most likely Interstitial Pneumonia with Autoimmune Features (IPAF). After receiving IVIg for five days, mycophenolate 1000 mg twice daily, and prednisone 60 mg daily, the patient was weaned to 3L O2 and discharged on mycophenolate 1500mg BID and prednisone 40 mg daily.
Discussion: IPAF is a relatively new diagnosis. As per the European Respiratory Society and American Thoracic Society in 2015, to be classified as IPAF, a disorder must show interstitial pneumonia with HRCT or surgical lung biopsy, be unable to meet Connective Tissue Disease (CTD) diagnostic criteria, exclude alternative etiologies, and satisfy criteria from two of the following three domains: clinical, serologic, and morphologic domain.1 Clinical criteria include physical manifestations of CTDs; serologic criteria include elevated levels of various auto-antibodies; and morphologic criteria include patterns suggested by HRCT or histologic patterns on surgical lung biopsy.1Our patient had significantly elevated RF and anti-CCP serum titers without any symptoms of defined CTD. Apart from her previous smoking history and remote history of jewelry washing, she had no environmental exposures or medication toxicity. HRCT showed extensive honeycombing and diffuse ground glass opacity representing either Usual Interstitial Pneumonia or fibrosing Nonspecific Interstitial Pneumonia. Therefore, she was given a diagnosis of IPAF.There is limited data on the pathophysiology, prognosis, and treatment of IPAF given its recent creation.1 Though associated with adverse effects in ILD, immunosuppression can benefit patients with CTD-associated ILD, and by extension IPAF.2 One large retrospective study found that mycophenolate improved FVC in IPAF patients.2-4 Most results suggest that patients with IPAF have decreased rates of mortality than those with ILD.1,5-6 Various studies suggest benefit from lung transplantation (LT) in patients with severe ILD, though there are no guidelines on LT in IPAF.7-8
Conclusions: This case highlights a poorly understood diagnosis. Our patient has shown minimal improvement with prednisone, mycophenolate, cyclophosphamide, and IVIg. There is still no explanation for the underlying mechanism behind IPAF. Given the limited data, further studies are needed to validate the appropriate treatment plan.