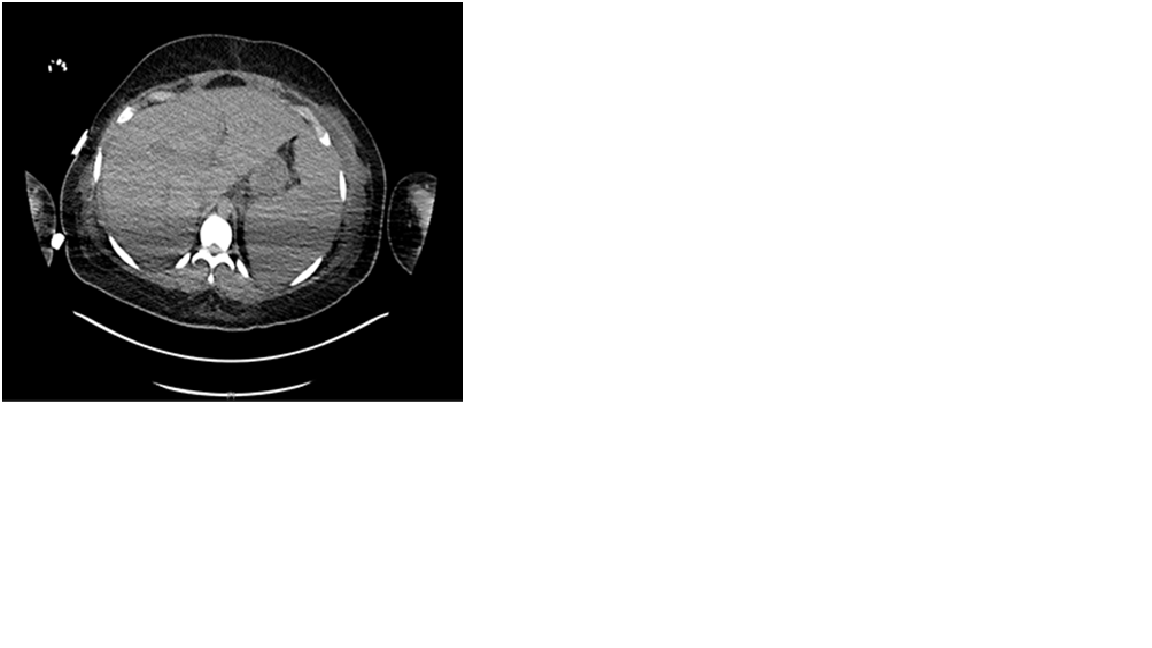
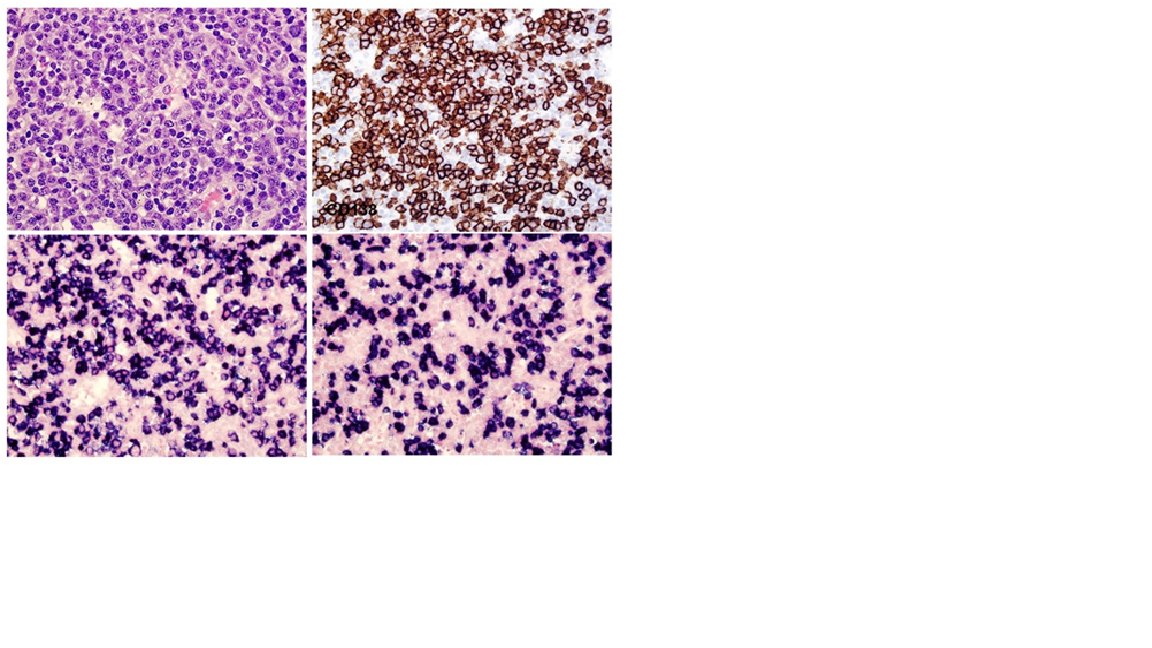
Case Presentation: A 22-year-old woman with a history of Adult Onset Still’s Disease presented with one week of acute abdominal pain, dyspnea and intermittent fevers. She was found to have thrombocytopenia to 15,000. Infectious workup was negative except for Rhinovirus/Enterovirus on viral panel. HIV and HHV8 were negative. MRI Abdomen showed hepatosplenomegaly and computed tomography was negative for pulmonary embolism but showed “pathologic left axillary and retropectoral adenopathy.” Her renal function worsened, requiring hemodialysis. Bone marrow biopsy showed hemophagocytosis, and a core needle lymph node biopsy showed focal histiocytes with hemophagocytosis and polytypic plasma cells. She was diagnosed with hemophagocytic lymphohistiocytosis (HLH) and treated with Anakinra and dexamethasone tapered over 8 weeks with resolution of renal failure. Two weeks after conclusion of the steroid taper, she presented with abdominal pain and edema. She was afebrile, tachycardic, and normotensive with normal oxygen level. Exam was notable for scleral icterus, peripheral edema, and diffuse abdominal tenderness. Labs showed thrombocytopenia, acute renal failure, cholestatic liver injury and coagulopathy. Cytokine and inflammatory markers were elevated, including IL-6, sIL-2 receptor, and VEGF. Workup for neoplasm by PETCT and peripheral flow cytometry were negative.An excisional axillary lymph node biopsy showed increased polytypic plasma cells without evidence of lymphoma, plasma cell neoplasm, or HHV8. She was treated with siltuximab for HHV8-negative idiopathic Multicentric Castleman disease (HHV8-iMCD) with recovery of platelets and renal function.
Discussion: Castleman Disease (CD) describes a group of lymphoproliferative disorders that share common histologic features, first described in 1956 in patients with benign localized lymph node enlargement (1). In the US, an estimated 6500 to 7700 cases of CD are diagnosed annually (2). MCD is thought to be a B-cell lymphoproliferative disorder (3). The systemic manifestations, driven by IL-6, include fever, night sweats, weight loss, hepatosplenomegaly, and volume overload (4). Multicentric disease differs from unicentric disease in that it involves multiple regions of lymphadenopathy, hepatosplenomegaly, cytopenias, and organ dysfunction (5). The disease is further categorized into HHV8 associated and HHV8 negative variants. HHV8 negative variants include POEMS-associated iMCD, and TAFRO syndrome iMCD. Management varies significantly based on disease subtype. The patient’s constellation of symptoms presented a diagnostic challenge requiring a coordinated multidisciplinary approach. The diagnosis of HHV8-iMCD was favored for several reasons. Her diffuse lymphadenopathy and hepatosplenomegaly in the setting of elevated cytokine/inflammatory markers were consistent with multicentric subtype. Periphery and lymph nodes were negative for HHV8. She did not have myelofibrosis to fulfill the diagnostic criteria for the TAFRO subtype of MCD. She did not have polyneuropathy, endocrinopathy, or monoclonal plasma cell disorder to fulfill the POEMs subtype. No evidence of neoplasm or lymphoma was identified. Therefore, treatment was initiated with anti-IL-6 therapy as the primary driver of her HHV8-iMCD.
Conclusions: This case highlights the challenge of diagnosing HHV8-iMCD. A multidisciplinary approach coordinated by hospital medicine was key to synthesizing this patient’s complex workup over multiple hospitalizations.