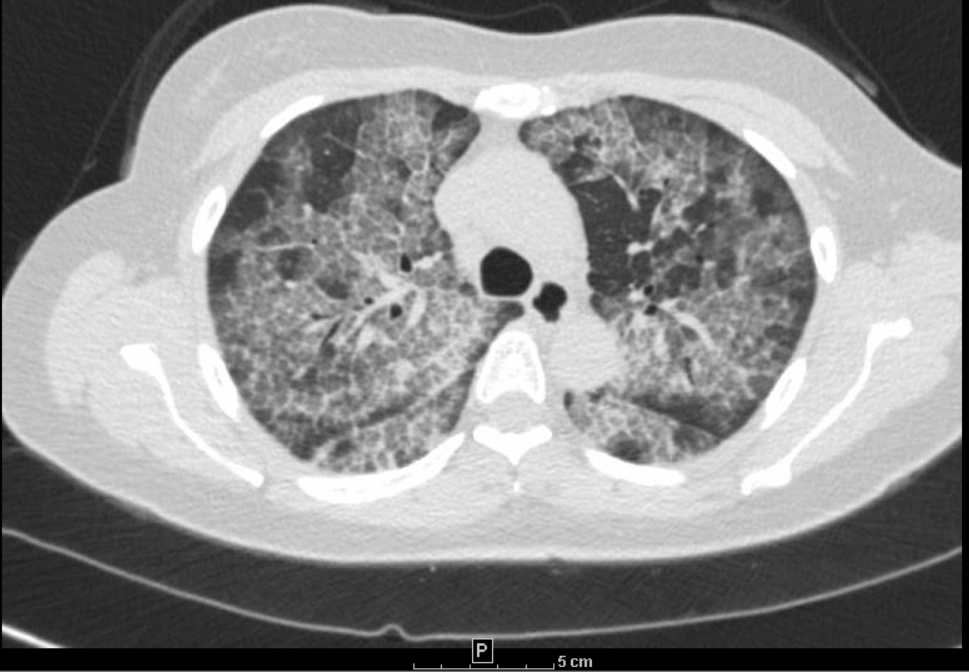
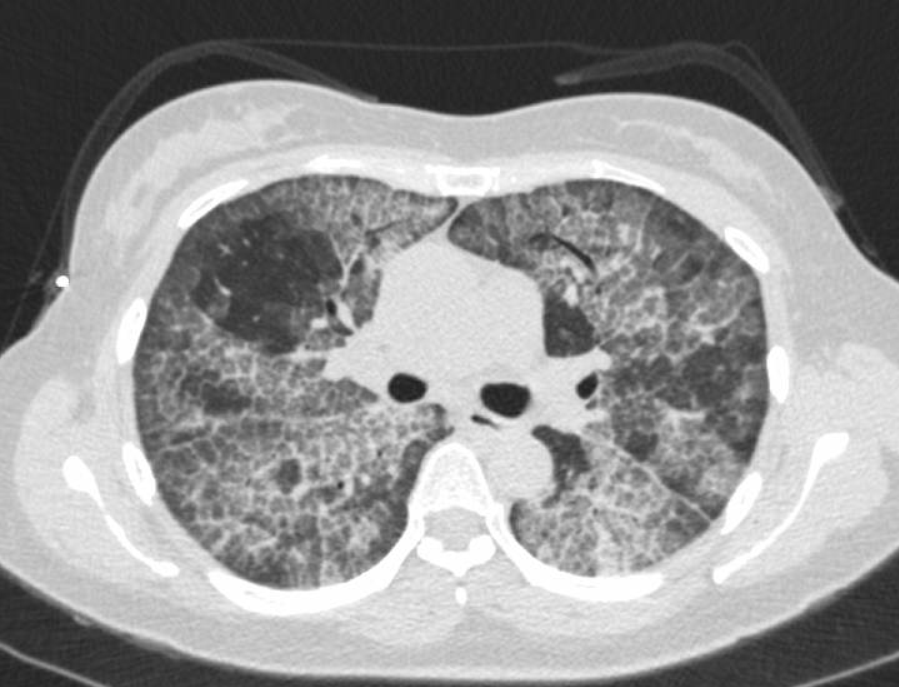
Case Presentation: A 43 year old Asian female with no significant past medical history presented to the hospital with complaints of cough and progressively worsening shortness of breath for 1 year. Patient denied any family history of lung diseases, nonsmoker, no significant smoke exposure, no recent travel. Patient works in a nail salon, and reports daily exposure to chemicals associated with her profession. On exam, vitals were normal apart from a resting oxygen saturation of 81% in room air, which improved to 97% with 2 L of oxygen via nasal cannula. The rest of her physical exam was unremarkable. CXR showed bilateral interstitial infiltrates. CT chest obtained showed crazy paving pattern suggestive of pulmonary alveolar proteinosis. Inflammatory markers, CRP and ESR were normal, Endo bronchial cultures were negative for infectious organisms. A high titer of serum anti-GM-CSF autoantibodies was noted. Other autoimmunity studies were negative. She underwent a bronchoscopy and broncho alveolar lavage and bronchial biopsies, with pathologic confirmation of protein alveolar proteinosis. She underwent serial whole lung lavage with improvement in oxygenation and symptoms.
Discussion: Pulmonary alveolar proteinosis (PAP) is a rare lung disorder characterized by alveolar accumulation with surfactant which impairs gas exchange leading to a severe hypoxemia and an increased risk of opportunistic respiratory infections. Etiology may be congenital (from mutations in surfactant proteins or granulocyte macrophage-colony stimulating factor (GM-CSF) receptor genes), secondary (from toxic inhalation or hematological disorders), and auto-immune (anti-GM-CSF antibodies blocking activation of alveolar macrophages), the latter accounting for 90% of cases. Patients typically present with cough and progressive exertional dyspnea which develops over weeks to months. Common examination findings include fine rales, clubbing and cyanosis. Radiologic findings often suggest diagnosis of PAP, which is confirmed by findings on a BAL specimen and endo bronchial biopsies. The most common treatment for patients with moderate to severe symptoms is a whole lung lavage. Alternative treatments include subcutaneous or inhaled granulocyte-macrophage colony-stimulating factor and lung transplant for severe cases refractory to treatment.
Conclusions: Pulmonary alveolar proteinosis (PAP) is a rare lung disorder, however clinicians should have a high index of suspicion for patients who present with chronic cough and bilateral ground glass interstitial infiltrates.