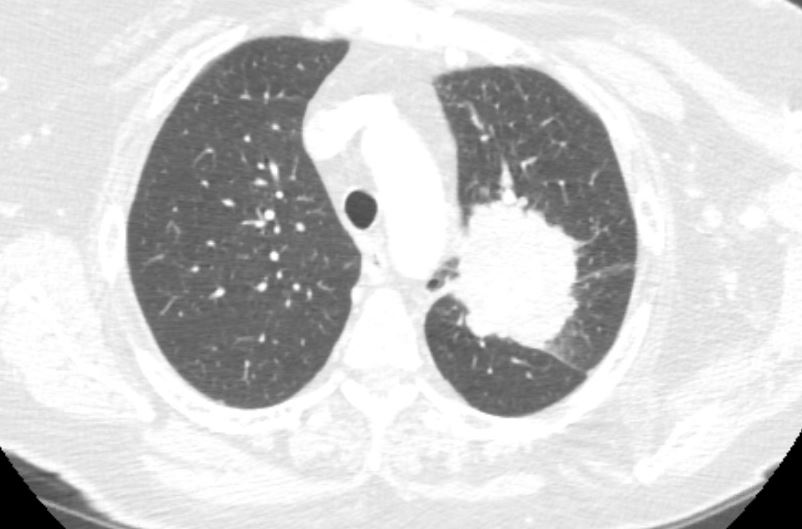
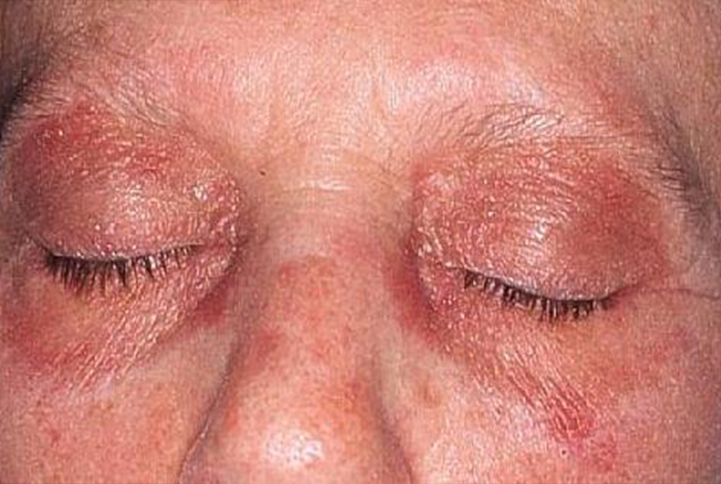
Case Presentation: A 65-year-old female with history of 70 pack years smoking, chronic hip pain, GERD, COPD presented to the ED with a 6 months complaints of diffuse pruritic skin rash associated with progressive facial swelling, fatigue and muscle weakness. She had been seen multiple times in the Dermatology clinic and punch biopsy revealed perifollicular and superficial perivascular inflammation. She was treated with topical and oral steroids, topical and oral anti-histamines without any relief. About a month prior to presentation she was seen by her primary care doctor for routine lung cancer screening and CT chest at that time showed a malignant process measuring 7.8 cm x 5.1 cm in the left lung with infiltration and encasement of the bronchovascular structures. On examination, BP 100/50 mmHg, HR 95 bpm, RR 16 bpm, and T 36.9 degrees Celsius. She had violaceous periorbital edematous erythema, slightly scaly around both eyes suspicious for Heliotrope rash, poikilodermatous erythema of neck, upper chest, entire back and slightly scaly erythematous plaques overlying MCPs and PIPS suspicious for Gottron’s papules. She was taken to the bronchoscopy suite for EBUS-TBNA of the left lung. Pathology revealed small cell lung carcinoma. In terms of her rash, it was diagnosed as DM presenting as a paraneoplastic syndrome in setting of small cell lung carcinoma. She was treated with intravenous steroids and transitioned to orals at discharge.
Discussion: Lung cancer, particularly small cell carcinoma is one of the most frequently associated with paraneoplastic syndromes due to its origin in neuroendocrine cell precursors. DM is an autoimmune idiopathic inflammatory myopathy associated with typical cutaneous manifestations. Characteristic cutaneous features include erythematous to violaceous papules over the extensor surfaces of MCP and/or PIP joints known as Gottron’s papules, and a violaceous periorbital erythema often accompanied by edema, referred to as heliotrope rash. The most common systemic symptom is proximal muscle weakness. The mechanisms by which neoplastic diseases generate DM are not fully understood. Classically, it has been considered as a humoral-mediated disease through the production of antibodies against tumor cells, which generates a cross reaction with one’s own muscle cells. Current evidence suggests that cellular-mediated immunity and alterations in the innate immune response plays a fundamental pathogenic role. Several studies have aimed to establish the profile of patients with a higher risk of presenting an underlying neoplasm. The presence of dysphagia, dyspnea, heliotrope erythema, muscle weakness, and arthralgia has been associated with an increased risk of malignancy. High clinical suspicion, imaging tests and histo-pathological studies are necessary to reach an adequate diagnosis.
Conclusions: DM is a disease with a relatively simple clinical diagnosis; however, clinicians should consider the possible association of DM with malignancy. The existence of occult primary tumors should be excluded in all patients with a diagnosis of DM, based on patient’s age, gender, local epidemiology and risk factors. Patients aged >40 years should be particularly screened for potential malignancies. Studies suggest that early diagnosis and timely treatment is crucial for the prognosis of the tumor. This case report underscores the importance of screening for malignancy in DM and aims to increase the clinical awareness of this disorder as a paraneoplastic phenomenon.