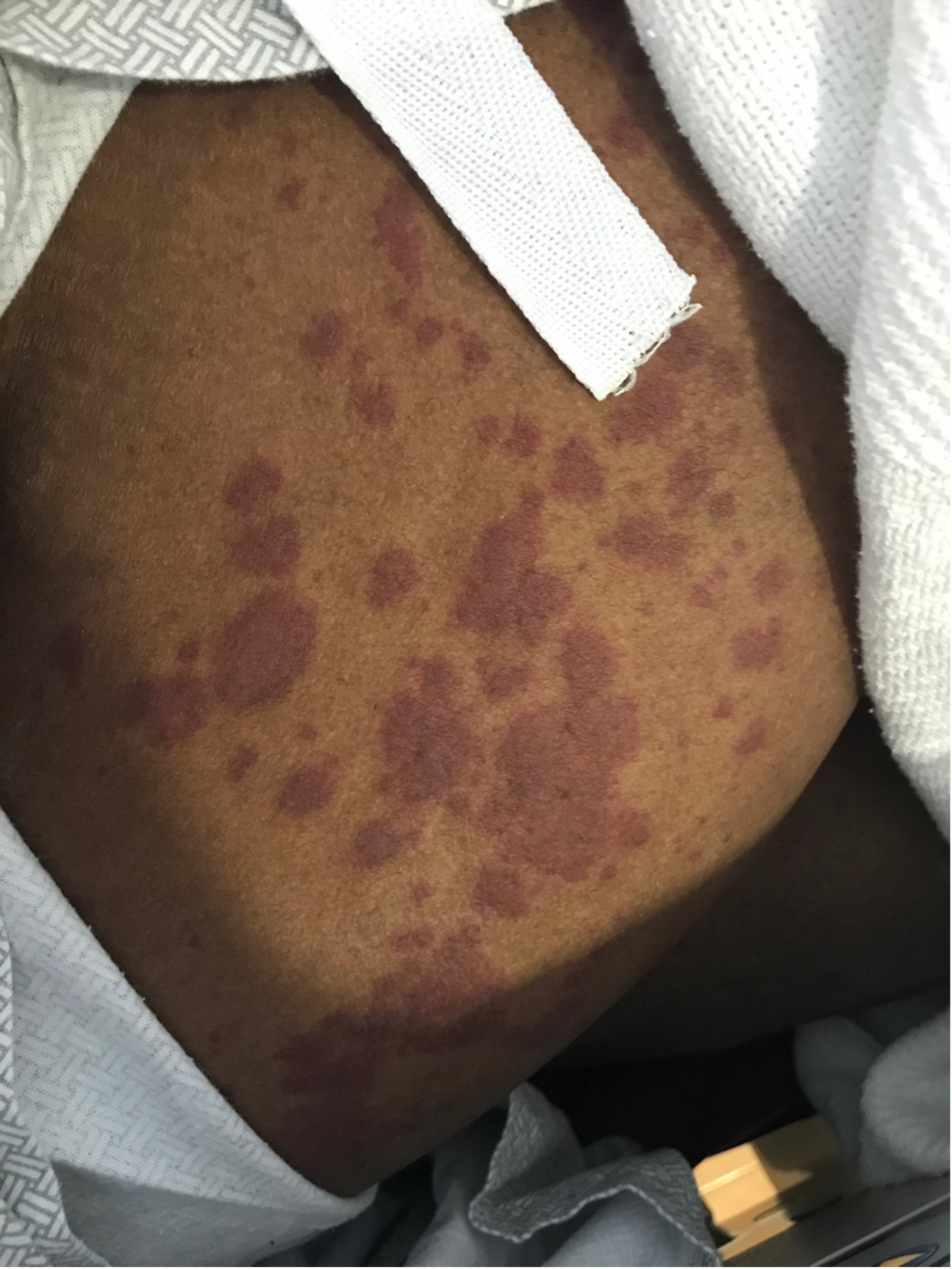
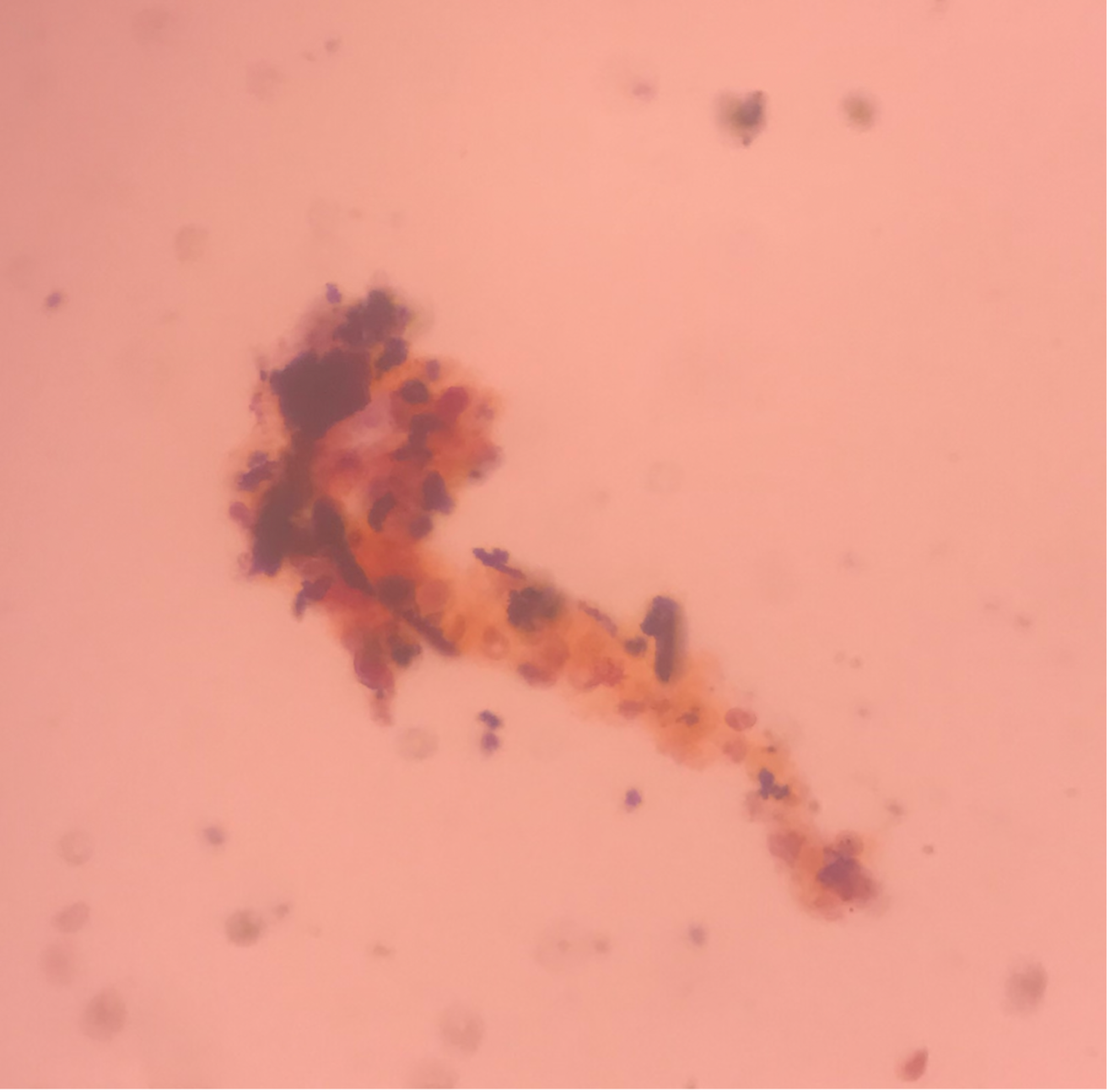
Case Presentation: A 29-year-old man with a history of morbid obesity, asthma, and chronic tracheostomy presented with 3 weeks of abdominal pain. The pain was intermittent and epigastric, without relationship to eating. He had poor appetite and nausea, though no emesis. He denied use of ibuprofen or alcohol. He denied fevers or chills. The pain worsened and began awakening him from sleep. He initially presented to an outside hospital. Basic labs including CBC, CMP, and lipase were unremarkable. RUQ ultrasound showed cholelithiasis without cholecystitis, and no bile ductal dilation. He was discharged home, but his abdominal pain became unremitting and constant. He then presented to our institution for further evaluation.Upon arrival, the patient’s abdominal exam was remarkable for nonlocalized tenderness to palpation of the upper abdomen. The patient was also noted to have a violaceous, non-blanching palpable rash (Figure 1), most prominent on the bilateral flanks and extending into the lower abdomen. Laboratory workup showed a hemoglobin of 11.7, platelets of 337, and creatinine of 1.22. ESR was elevated to 36 and c-reactive protein was elevated to 42.7. Urinalysis showed 2+ protein, 3+ blood, 0 WBC, and >50 RBCs. Urine protein/creatinine ratio was 683.5 mg/g. Urine sediment review showed red blood cell casts (Figure 2). HIV, hepatitis panel, cryoglobulins, ANCA and anti-GBM antibodies were negative. CT abdomen/pelvis with contrast showed stranding and fluid tracking from the second and third portion of the duodenum. A right flank skin punch biopsy showed sparse perivascular and interstitial neutrophilic infiltrate with cellular debris, purpura, and scattered eosinophils, consistent with leukocytoclastic vasculitis. Given the purpura, abdominal pain and renal involvement, a diagnosis of immunoglobulin A vasculitis was made. He was initiated on prednisone, with immediate improvement in his abdominal pain. He was discharged and follow-up revealed complete resolution of his GI symptoms and rash.
Discussion: IgA vasculitis (IgAV) is the most common systemic vasculitis in the pediatric population with an incidence of 3 to 27 per 100,000. IgAV is less common in adults, with an incidence of 0.8 to 5.1 per 100,000 and has been associated with a more severe presentation. The classic constellation of findings includes palpable purpura without thrombocytopenia, arthralgia, abdominal pain, and renal disease. Pathogenesis is mediated by IgA immune complex deposition. Biopsies of IgAV typically reveal leukocytoclastic vasculitis primarily affecting the small superficial vessels. Diagnosis is made with a combination of clinical and histopathologic data. The European Alliance of Associations for Rheumatology (EULAR) classification criteria, which requires the presence of purpura and at least 1 of 4 criteria including abdominal pain, histopathology, arthritis and renal involvement, has been found to have high sensitivity and specificity for diagnosis in the adult population. In this case, the diagnosis was delayed due to atypical features, including the patient’s age, body habitus limiting prior evaluations, and difficulty communicating due to presence of a tracheostomy.
Conclusions: IgA vasculitis can occur in the adult population. Careful consideration of unusual diagnoses in atypical patient demographics, and synthesis of history and physical exam with laboratory and pathologic data, is needed to arrive at the correct diagnosis and management.