Case Presentation: A 65-year-old Japanese man presented with a 5-month history of bilateral leg edema. He visited his family doctor 3 months ago and started taking furosemide, but the symptom didn’t improve. His past medical history included chronic atrial fibrillation. He reported malaise, constipation and weight gain but denied fever or night sweats. On admission, he was ill-appearing and afebrile. The other vital signs were normal. The abdominal examination revealed splenomegaly. There was bilateral leg edema. In laboratory testing, there was a mild elevation of transaminases (AST 95 IU/L, ALT 55 IU/L) and markedly elevated of LDH 1430 IU/L. TSH was 0.36 µU/ml and free-T4 0.49 ng/dl. The peripheral blood film showed no abnormality.
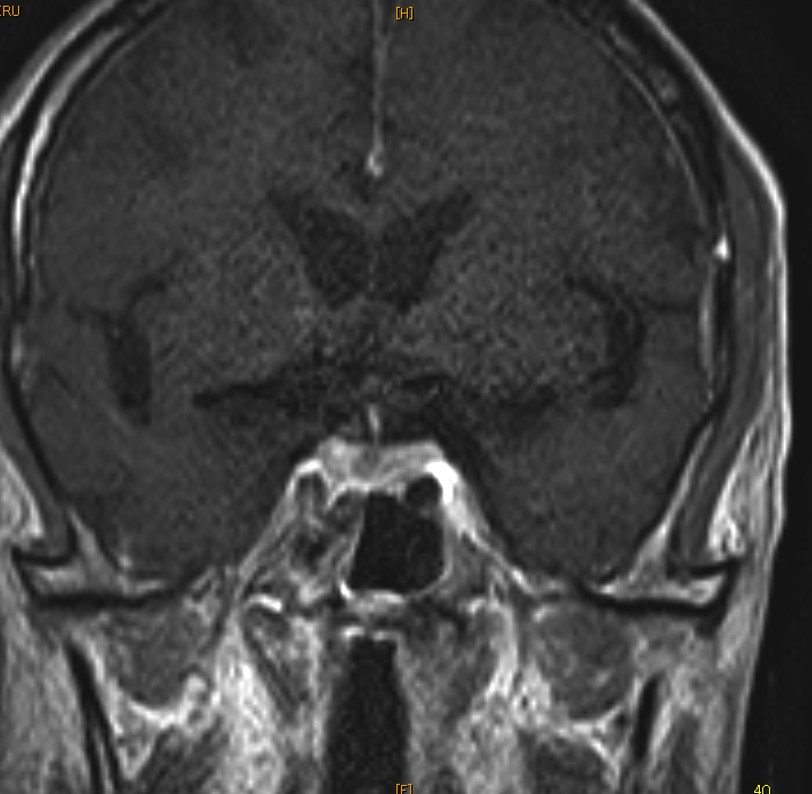
Initial differential diagnoses included hypopituitarism or non-thyroidal illness. Additional testing suggested hypopituitarism with somatomedin C 30 ng/ml, LH <0.10 mIU/ml and FSH 0.26 mIU/ml. The serum ACTH and cortisol were within normal limits. There is no response of TSH by TRH stimulation test. Contrast-enhanced brain MRI showed a mass-like lesion with the filling defect in the pituitary gland (Figure 1). Oral levothyroxine was initiated. Because of the high LDH level and splenomegaly, a possibility of malignant lymphoma was considered but a whole-body contrast-enhanced CT showed no lymphadenopathy. On the seventh day of hospitalization, he developed hypotensive and pancytopenia and died despite attempts of cardiopulmonary resuscitation.
The autopsy showed there was infiltration of CD20-positive abnormal lymphocytes in the anterior lobe of the pituitary gland. In addition, the abnormal cells were seeded into blood vessels of organs including skin, small intestine, and bone marrow. Autopsy diagnosis of intravascular lymphoma (IVL) was made. The cause of hypopituitarism was considered pituitary destruction by IVL cells.
Discussion: Anterior hypopituitarism caused by IVL is relatively rare but can be a rapidly fatal condition. IVL is characterized by dissemination of lymphoma cells within the lumen of blood vessels in the entire body. Patients with IVL occasionally present with nonspecific symptoms and thus mimic other benign diseases. The diagnosis of IVL can be very challenging and it requires tissue diagnoses by random skin biopsy, or biopsy of bone marrow, brain or other organs. However, if conditions are rapidly aggressive as in this case, patients with IVL are often diagnosed in an autopsy.
As a cause of hypopituitarism, there were only less than 20 reported cases of IVL and most of the cases were diagnosed by autopsy. If there would be no history of prior trauma or radiotherapy but findings suggestive of malignant lymphoma such as high LDH level and splenomegaly, IVL should be considered even if image testing showed no lymphadenopathy. Life-saving chemotherapy, R-CHOP, can be indicated for some patients. As a hospitalist, we must know the importance of diagnosing and managing this disease although it may challenge us with diagnostic difficulty.
Conclusions: IVL is a rare but potentially curable cause of hypopituitarism. We should take into consideration it if there are splenomegaly and high LDH level and no other obvious causes of hypopituitarism. A diagnosis of IVL can be challenging and tissue biopsy should be performed swiftly.