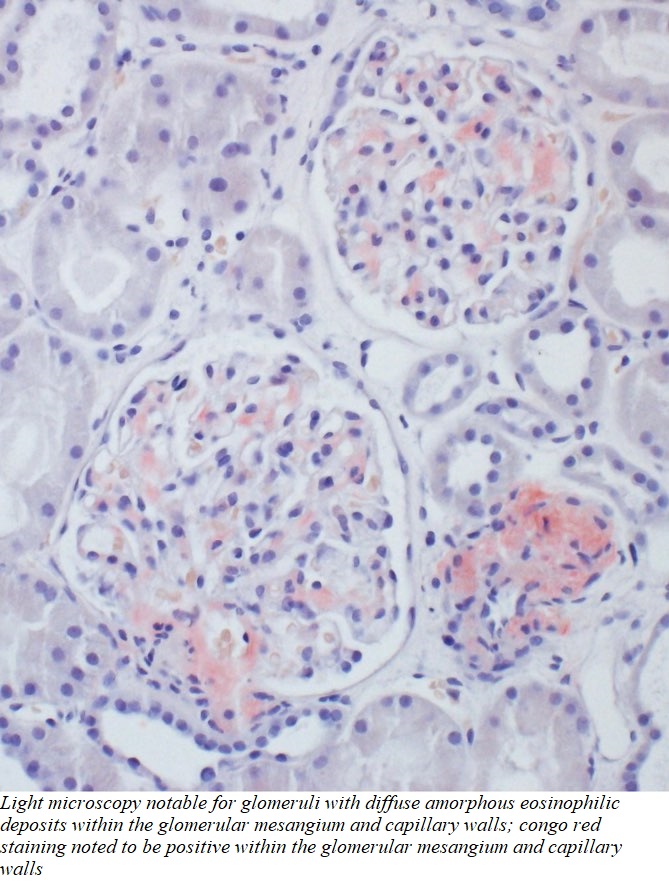
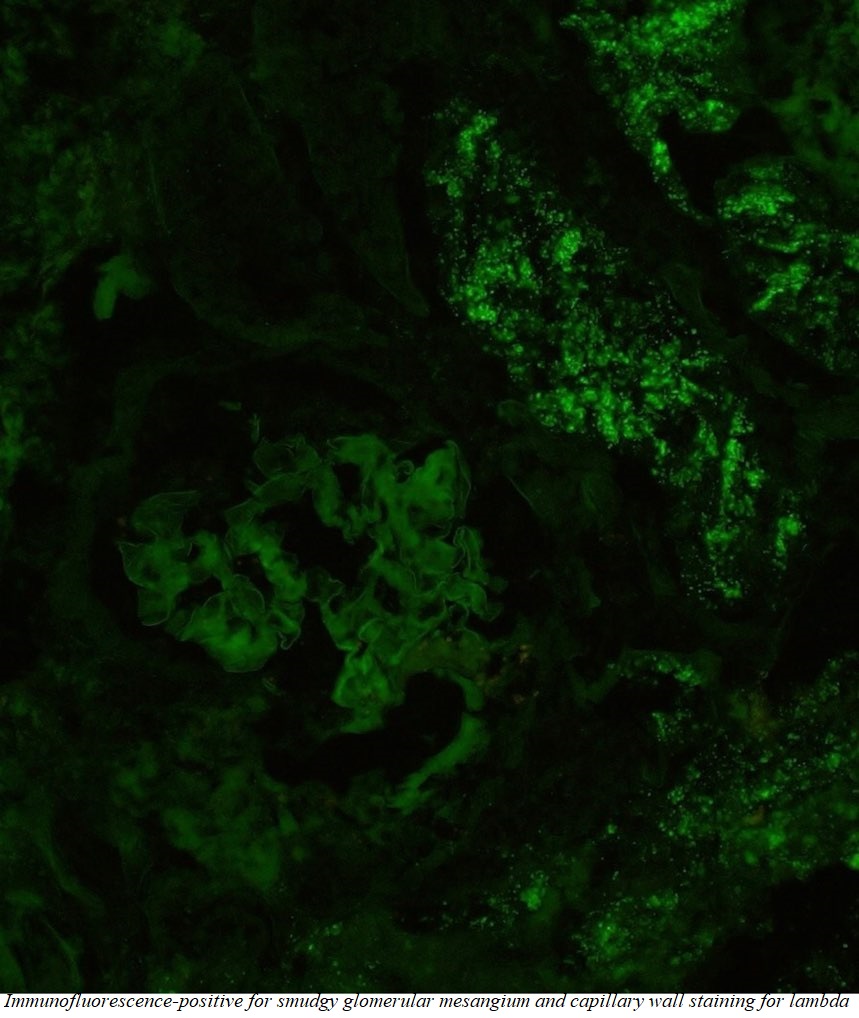
Case Presentation: An elderly male with a pertinent past medical history of carpal tunnel syndrome, and chronic diarrhea, presents with complaints of progressively worsening anasarca and associated dyspnea. He has had multiple hospitalizations for dyspnea over the last six months, thought to be due to transudative pleural effusions. Outpatient work-up, including echocardiogram and subspecialty consultation with Cardiology, have been unrevealing, with anasarca being attributed to non-cardiac causes, initially managed with diuresis. Given the generalized distribution of edema, a workup was initiated to evaluate for the etiology of anasarca. Medications, diet/nutritional status, and cardiac causes were ruled out as contributing factors. Lab studies revealed elevated transaminases which were further worked up with ultrasound of the liver given new-onset abdominal pain during the hospital course, which ultimately revealed chronic cholecystitis as well as hepatomegaly. Urine studies were notable for nephrotic range proteinuria, which complemented the hypoalbuminemia, edema, and renal dysfunction to suggest the culprit for our patient’s anasarca were the kidneys. Primary and secondary causes of nephrotic syndrome were investigated, leading to a renal biopsy which was notable for mesangial expansion by presence of abundant, small, overlapping, non-branching fibrils morphologically compatible with amyloid. Light microscopy was notable for glomeruli with diffuse amorphous eosinophilic deposits within the glomerular mesangium and capillary walls; Congo red staining noted to be positive within the described glomerular and vascular wall deposits; polarization microscopy confirmed apple green birefringence. Immunofluorescence was positive for smudgy glomerular mesangial and capillary wall staining for lambda. A diagnosis of AL-type Amyloidosis was made.
Discussion: Amyloidosis is a term representing a disease process that is the result of extracellular deposition of fibrils formed by proteins in a beta-sheet configuration, called amyloid. Amyloid is resistant to proteolysis and hence, accumulates in tissues, leading to interruption in its respective function. The clinical manifestations noted in Amyloidosis are generally guided by the substrate protein, amount of amyloid and the location of deposition. In this case, features that were suggestive of amyloidosis included the physical appearance of the patient (prominent facial features and body structure), anasarca initially thought to be of cardiac origin, signs of cardiac conduction abnormalities, nephrotic-range proteinuria, hypoalbuminemia, as well as neuropathic symptoms. Of all the described features, the earliest clinical presentation of amyloidosis in this case was likely neuropathy diagnosed as carpal tunnel syndrome, merely a year prior.
Conclusions: AL-Amyloidosis is a rare systemic disorder with a rapidly progressive clinical course. It presents as a diverse and unique clinical picture, which can include nephrotic-range proteinuria, edema, cardiac abnormalities, hepatosplenomegaly, and carpal tunnel syndrome. This case is a good example of a patient with a variety of symptoms that when viewed individually can present as a collage of varied pathologies, but when analyzed as a collective, they are best explained by a single disease process. As diagnosticians, this reinforces the importance of a thorough history and tying the clinical presentation to the pathophysiology of the disease process we intend to treat.