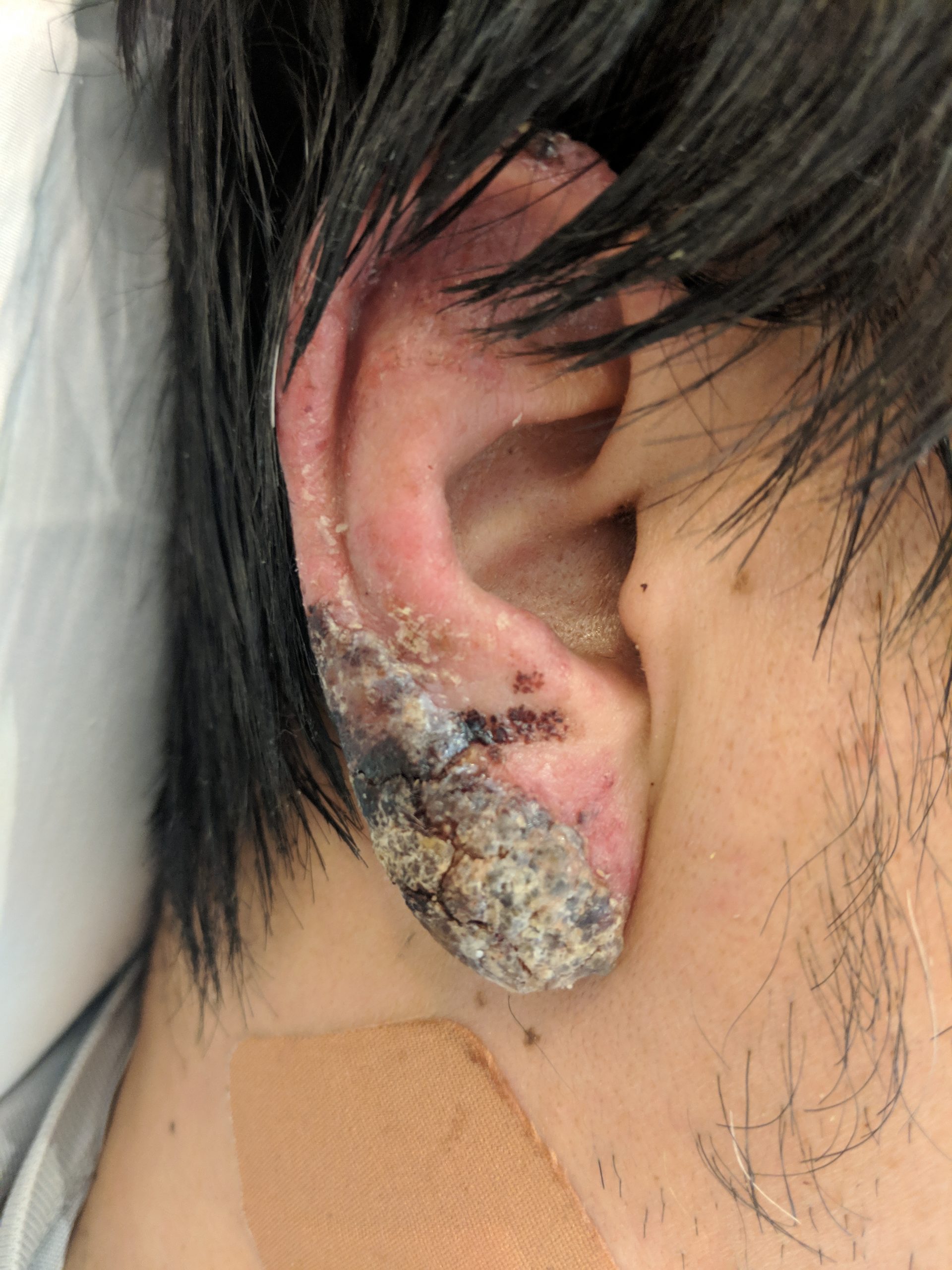
Case Presentation: A 43-year-old Japanese man without significant past medical history presented with one month of progressive malaise, fevers, chills, and swollen, painful lymph nodes on the right side of his neck. Prior to coming to our hospital, he was seen in an emergency department and was given a course of penicillin for presumed adenitis without improvement. 3 days prior to hospitalization, he also began to develop a painless, red, crusting rash on both of his ears, prompting his visit to our institution. He denied travel history, though he was born and raised in Japan. His exam was notable for a temperature of 101.6°F, blood pressure of 90/57 mm Hg, an erythematous malar-distributed plaque, erythema with skin breakdown and golden crusting over both ear lobes, faint erythematous macules on both thighs, and tender, mobile, enlarged, right cervical lymph nodes. His labs demonstrated normocytic anemia (11.7), thrombocytopenia (129), and elevation of aspartate aminotransferase (238), alanine aminotransferase (299), and alkaline phosphatase (217), with a c-reactive peptide of 3. An extensive infectious workup was unremarkable, prompting an excisional cervical lymph node biopsy that demonstrated histiocytic necrotizing lymphadenitis, and a skin biopsy of the ear that demonstrated a cytotoxic interface reaction with lymphocytic infiltrate. Rheumatology and dermatology felt strongly that the clinical picture was most consistent with Kikuchi-Fujimoto disease (KFD) with cutaneous vasculitis. Given the unusual and potentially disfiguring presentation, the patient was started on prednisone 20 mg daily and hydroxychloroquine 200 mg daily, with subsequent improvement at a 4-month follow-up.
Discussion: KFD, otherwise known as histiocytic necrotizing lymphadenitis, is a rare, benign, and self-limited process felt to be associated with certain viral infections. However, there have been case reports of KFD presenting with necrotizing vasculitis. KFD is seen more commonly among Asian ethnicities (particularly those of Japanese descent), described as a benign, subacute lymphadenitis with associated systemic symptoms. The etiology is believed to be due to an inciting viral infection, with symptoms developing subsequently, and resolving slowly over weeks to months. The diagnosis is usually made microscopically, with biopsy of affected lymph nodes characterized by histiocytic necrotizing lymphadenitis. Although most cases of KFD are self-limited, substantial and potentially problematic presentations including vasculitis have been reported, invariably requiring hospitalization. For localized disease, symptomatic relief is usually sufficient, though in generalized or troublesome sites of disease manifestation, systemic immunosuppression is warranted. Disease recurrence or development of systemic lupus erythematosus have been reported, so continued monitoring is usually warranted. Despite the low incidence of KFD, it is nevertheless an important differential consideration of a patient such as ours. Inappropriate focus on mimics of KFD can lead to prolonged hospitalizations and overuse of invasive testing, causing undue physical and financial burden to both patients and the healthcare system.
Conclusions: KFD is a rare and difficult diagnosis to make but should be considered in a patient presenting with fevers and localized lymphadenopathy, especially in Asian ethnicities. Early lymph node biopsy is essential to the diagnosis and ultimate management.