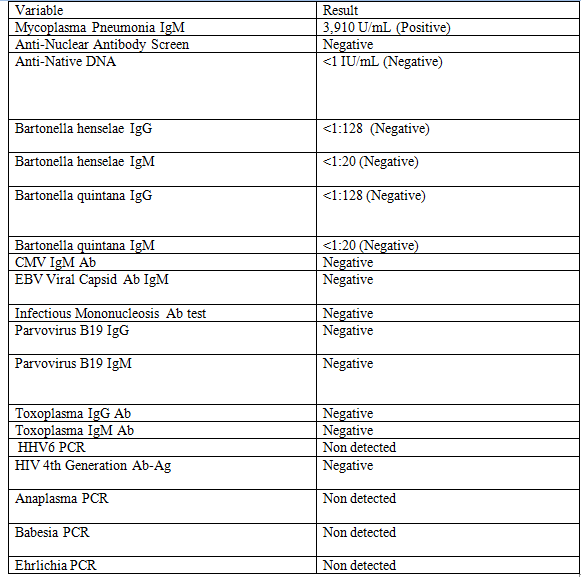
Case Presentation: A 9-year-old boy with sickle cell disease (SCD) presented with 5 days of fever and left-sided neck pain. His physical exam was notable for tender cervical lymphadenopathy bilaterally as well as objective fever. Laboratory studies revealed leukopenia, a microcytic anemia, and elevated c-reactive protein. A nasopharyngeal viral PCR panel was negative. An ultrasound of the neck was obtained, which showed bilateral cervical lymphadenitis. Following blood cultures and the administration of IV antibiotics, he was admitted to the floor. Despite broad spectrum antibiotic therapy, the patient’s condition did not improve over the next week. A more extensive workup (Figure 1) was undertaken with the only notable result being a positive Mycoplasma pneumoniae IgM, for which azithromycin was started. He continued to remain febrile with left sided cervical lympadenopathy, and elevated inflammatory markers. With input from infectious disease, hematology/oncology and surgery, as well as the patient’s mother, the decision was made to perform an excisional biopsy of his largest cervical lymph node. Pathology revealed morphologic features suggestive of histiocytic necrotizing lymphadenitis, a pattern most typical of Kikuchi-Fujimoto disease (KFD). Prednisone was started resulting in symptomatic improvement and the patient was discharged home after an otherwise uncomplicated hospital course.
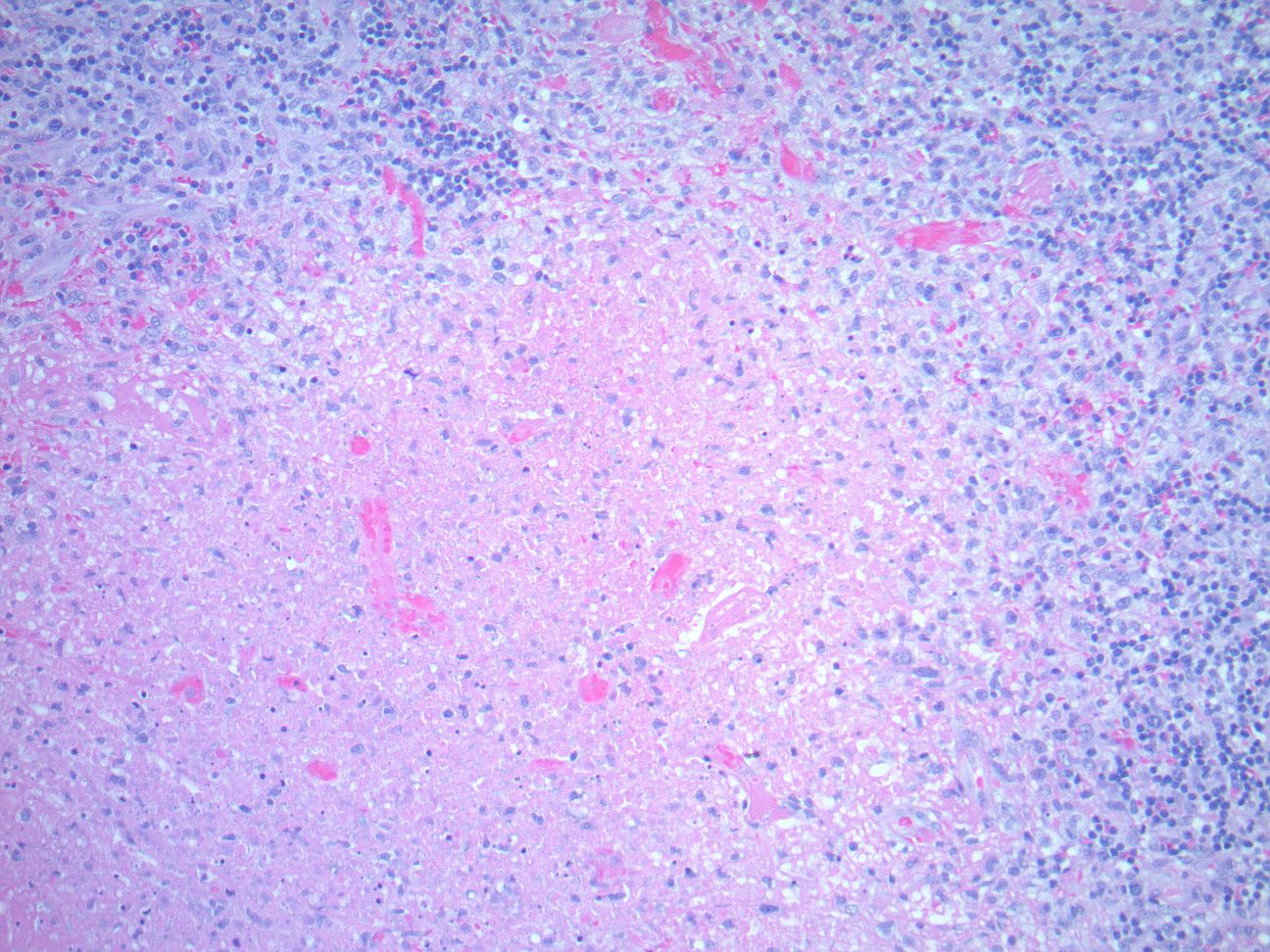
Discussion: KFD is a characterized clinically by painful cervical lymphadenopathy, occasionally associated with fever, malaise and upper respiratory symptoms. Laboratory evaluation is often nonspecific with a variety of findings being described including anemia, leukopenia and elevated inflammatory markers. While the pathogenesis is unknown a hyperimmune response in genetically susceptible patients has been proposed. There are reported cases of KFD in SCD patients with a number of them being complicated by the development of autoimmune disorders. The diagnosis of KFD is based on lymph node histology (Figure 2) and can be difficult given the nonspecific signs and symptoms. Some patients have gone on to develop systemic lupus, however the majority have a benign clinical course. In this case, the patient showed no improvement despite antibiotic treatment. His lymphadenopathy, ongoing fevers, and laboratory abnormalities, as well as his underlying SCD, led the team to consider alternative diagnoses and move ahead with the appropriate diagnostic test.
Conclusions: This case demonstrates an example of a rare disorder, KFD, and also contributes to the small literature base describing KFD in patients with SCD. It also highlights the importance of revisiting a patient’s differential diagnosis and rearranging it based on the response to treatment. KFD is typically diagnosed weeks to months after symptom onset. In this case, the diagnosis was made during the initial hospitalization as the patient’s medical history, persistent symptoms, laboratory abnormalities and lack of response to antibiotics prompted further investigation.