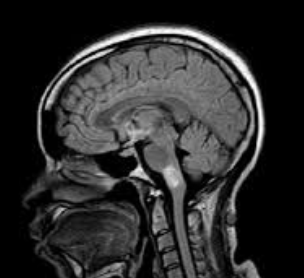
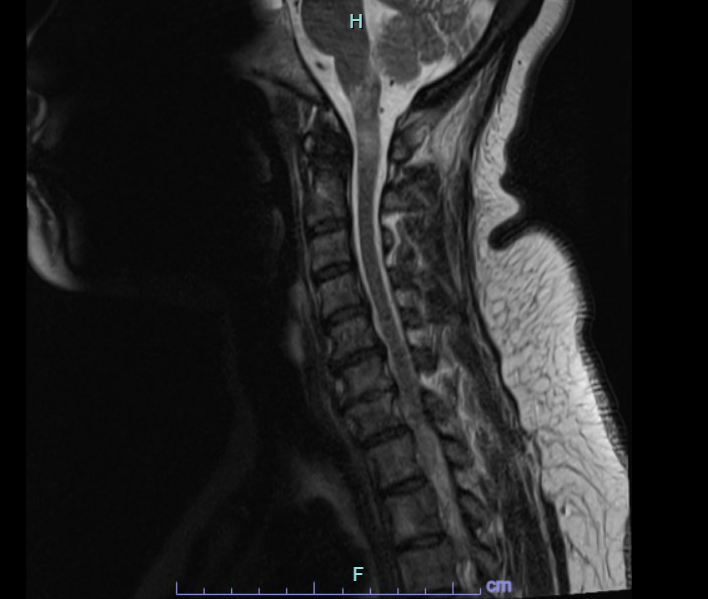
Case Presentation: A 47-year-old female with no significant past medical history presented to the emergency department in December with gastroparesis followed by progressive asymmetric weakness, numbness, and impaired gait associated with fall two years ago. MRI Brain was performed and unremarkable. She then developed a constant spinning sensation. She tried meclizine without benefit. The following month, she presented with numbness and tingling of the right hand that over the course of weeks spread to her left hand. She also described a “bandlike” thoracic sensation associated with upper/lower extremity weakness and balance impairment leading to fall. She was found to have an enhancing brainstem lesion on MRI brain performed on subsequent admission. MRI of the cervical/thoracic spine was consistent with demyelinating disease. She was treated with pulse IV Solu-medrol 500 mg BID for 3 days for central nervous system (CNS) demyelinating disease and then started on prednisone 80 mg daily. Myelin Oligodendrocyte Glycoprotein (MOG) antibody returned positive in serum. Also, found to have positive SSA/SSB without associated sicca or other clinical features. Given the constellation of recurrent short-segment myelitis, serum MOG antibody positivity and prominent therapeutic response to and dependence on steroids, the diagnosis of Myelin oligodendrocyte glycoprotein antibody-associated disorder (MOGAD) was made. Labs revealed negative Oligoclonal bands (OCB), anti-AQP4 neg, and + anti-MOG. IR-guided Lumbar puncture revealed: no white blood cells, few red blood cells, no xanthochromia, normal protein, and negative OCB. Borderline dsDNA noted. Patient is currently off steroids and receiving Rituximab-abbs (Truxima) infusions with neurology; other medications include Lacosamide and Cymbalta.
Discussion: Our patient case, along with an increasing number of reports, suggests a proportion of MOGAD with initial phenotypes suggestive of Multiple Sclerosis (MS). The presence of lesions with shapes and distributions compatible with MS lead to the misdiagnosis of MS. However, her serum MOG antibody seropositivity (cell-based assay) and a substantial dependency on steroids all supported the final diagnosis of MOGAD. Though considered as distinct disorders, manifestations of MS and MOGAD may overlap. Among them, short-segment myelitis constitutes the initial presentation in more than half of MS and up to 53% of MOGAD. In addition, around 6% to 17% of MOGAD patients have positive CSF OCB. Also, given their overlapping presentations, differentiation between the two disorders is extremely important based mainly on two reasons: 1) maintenance therapy of MS and MOGAD differs greatly: on the one hand, disease-modifying therapy (DMT) are mainly used in patients with MS; on the other, patients with MOGAD are particularly responsive to antibody-depleting treatments, such as Truxima, but may even deteriorate with DMT. 2) MOGAD is associated with a high risk of flare-ups after cessation of steroid treatment and may thus require close monitoring and careful steroid tapering at least over 6 months.
Conclusions: In conclusion, our case highlights the importance in differentiating between MS and MOGAD in patients with recurrent short-segment myelitis, especially with therapeutic response to steroids. Serum MOG antibody testing would aid in the correct diagnosis and initiation of appropriate treatment for MOGAD.